| |
T-20 Safety & Side Effects in TORO 1-2
|
| |
| |
Safety of Enfuvirtide in Combination With an Optimized Background of Antiretrovirals in Treatment-Experienced HIV-1-Infected Adults Over 48 Weeks
JAIDS Journal of Acquired Immune Deficiency Syndromes: Volume 40(4) 1 December 2005 pp 413-421
Trottier, Benoit MD*; Salgo, Miklos P MD, PhD###
From the *Clinique Medicale l'Actuel, Montreal, Quebec, Canada; Roche, Basel, Switzerland.
Abstract
Background: Antiretroviral tolerability is a critical factor contributing to treatment outcome. The T-20 Versus Optimized Background Regimen Only (TORO) studies assessed the safety and efficacy of enfuvirtide in treatment-experienced HIV-1-infected patients.
Methods: A total of 997 patients were randomized at a 2:1 ratio to an optimized background antiretroviral regimen plus enfuvirtide (n = 663) or an optimized background regimen alone (control group; n = 334). Control patients could switch to enfuvirtide on virologic failure.
Results: In total, 26.5% of patients randomized to enfuvirtide and 36.6% to the control group discontinued study treatment before week 48; the percentage of patients withdrawn for safety reasons (including adverse events [AEs], deaths, and laboratory abnormalities) was 14.0% in the enfuvirtide group and 11.6% in the control group. Injection site reactions (ISRs) occurred in 98% of enfuvirtide patients and led to treatment discontinuation in 4.4%. Treatment-related (defined as possibly, probably, or remotely) AE rates per 100 patient-years were lower with enfuvirtide (96.2) than in the control group (149.9); diarrhea, nausea, and fatigue, the most frequently reported AEs, were significantly less frequent with enfuvirtide than in the control group. Pneumonia was significantly more frequent in patients treated with enfuvirtide (6.7 vs. 0.6 events per 100 patient-years), although the incidence was within expected ranges for this population. Lymphadenopathy was also higher in enfuvirtide-treated patients (7.1 vs. 1.2 events per 100 patient-years) for control patients.
Conclusion: The addition of enfuvirtide to an optimized background regimen does not exacerbate AEs commonly associated with antiretrovirals. ISRs limited treatment in <5% of patients.
AUTHOR DISCUSSION
Results of the combined analyses of the TORO trials confirm the safety profile and tolerability of enfuvirtide with an optimized background antiretroviral regimen over 48 weeks in this multidrug-resistant, treatment-experienced, HIV-1-infected population. Derived from a study population of nearly 1000 patients, these data provide reassurance that the addition of enfuvirtide to an optimized background regimen does not exacerbate the AEs commonly associated with antiretroviral agents in the background regimen, especially gastrointestinal AEs. The overall safety profile documented at week 24 is maintained at week 48. When increases in AEs were observed with enfuvirtide, they were generally considered to be clinically manageable, with little impact on the overall assessment of benefit and risk of this treatment.
The study design resulted in a marked difference in the extent of exposure between treatment groups. The incidence density (number of patients with an event per 100 patient-years of study treatment exposure) for each AE throughout the first 48 weeks of the treatment period was therefore identified as an appropriate reporting methodology, because it takes into account these differences and provides an assessment of AEs that reflects the extent of exposure to the studied treatment regimens.
Overall, the rate of AEs observed in this study population was quite high, but this was expected, given the advanced stage of disease and level of treatment experience of this patient population. The types of AEs that were reported are generally consistent with what would be expected in such patients. It is of note that 3 of the most commonly reported AEs in this study (all causality, excluding ISRs) occurred significantly less frequently in patients taking enfuvirtide: diarrhea (RR = 0.51, 95% CI: 0.40 to 0.63), nausea (RR = 0.52, 95% CI: 0.40 to 0.69), and fatigue (RR = 0.66, 95% CI: 0.49 to 0.90). The greater control of viral replication and immune recovery associated with enfuvirtide treatment may have contributed to this finding. Rates of other AEs favored enfuvirtide or were generally comparable between treatments.
Aside from the increase in treatment-emergent eosinophilia, which was not associated with any clinical events of hypersensitivity, laboratory abnormalities were comparable across treatments. No association was found with any lipid or glycemic laboratory parameters.
ISRs were the most common AEs associated with enfuvirtide and were seen in almost all patients. Most of these were mild to moderate. Over the 48-week study period, 11% of patients experienced severe pain that required prescription drugs, necessitated nontopical analgesics, or limited usual activities, but the rate of discontinuation attributable to ISRs remained low (<5% patients). A trend toward more severe ISRs in those with a lower body mass index has recently been confirmed. The severity of ISRs was found to be lower in patients with more peripheral fat, possibly because it is easier to avoid inadvertent intramuscular injections if the needle goes too deep.13 Because of the high incidence of these reactions, physicians should become familiar with the clinicopathologic correlation of these reactions before caring for HIV-1-infected patients undergoing this new mode of therapy.14
With regard to the lymphadenopathy (all causality) seen more frequently in patients treated with enfuvirtide compared with the control group, it is possible that enfuvirtide may have a certain degree of immunogenicity as indicated by the local reaction at the site of injection. Peripheral lymphadenopathy is a typical reaction after subcutaneous injection of immunogenic substances. Thus, the lymphadenopathy observed in subjects treated with enfuvirtide may possibly be attributable to the immunogenic properties of enfuvirtide and the large (90 mg twice daily) amount of enfuvirtide administered. Whether or not enfuvirtide is immunogenic in human beings has been difficult to determine, because more than 95% of HIV-infected patients in the pooled TORO trials had antibodies to gp41, which cross-reacts with enfuvirtide, before enfuvirtide treatment was initiated. No apparent differences were observed in the AE profile of enfuvirtide as a function of gp41 antibody status at baseline, however, and there was no evidence that increased antibody levels were associated with increased toxicities.15 Because the least-squares mean increase from baseline in CD4 cell count in the enfuvirtide group was approximately double that in the control group, some degree of immune reconstitution could also have contributed to the lymphadenopathy.
Systemic hypersensitivity reactions attributed to enfuvirtide were reported in a small number of patients (<1%) and recurred on rechallenge in some cases. Events included, in various combinations, rash, fever, nausea and vomiting, chills, rigors, hypotension, and elevated serum liver transaminases and may include primary immune complex reaction, respiratory distress, and glomerulonephritis. Patients developing signs and symptoms suggestive of a systemic hypersensitivity reaction should discontinue enfuvirtide treatment and seek medical evaluation immediately. Therapy with enfuvirtide should not be restarted after systemic signs and symptoms consistent with a hypersensitivity reaction considered to be related to enfuvirtide. Risk factors that may predict the occurrence or severity of hypersensitivity to enfuvirtide have not been identified.
An increased rate of *pneumonia was observed in patients treated with enfuvirtide compared with the control arm. The rates of pneumonia reported in the literature from the pre-HAART era for HIV-infected patients are 5 to 9 events per 100 patient-years.16-20 More current data from the Adult Spectrum of Disease Study (Seattle, WA) show a lowering of the rate of pneumonia in HIV-infected patients with the advent of HAART use, with rates of bacterial pneumonia as captured in that study of 10.9 to 27.0 per 100 patient-years in the pre-HAART era, decreasing to 6.5 to 12.5 per 100 patient-years in the HAART era for patients with CD4 counts of 101 to 200 cells/mm3 and 0 to 100 cells/mm3, respectively. Including bacterial pneumonia and the term pneumonia nos, rates were 17.8 to 41.4 per 100 patient-years in the pre-HAART era, declining to 10.3 to 20.1 per 100 patient-years in the HAART era for patients with CD4 counts of 101 to 200 cells/mm3 and 0 to 100 cells/mm3, respectively (data on file, Roche). The incidence observed in the combined enfuvirtide patient group (6.7 events per 100 patient-years of exposure) was thus within the range reported for this patient population, whereas the incidence of *pneumonia seen in the control group (0.6 events per 100 patient-years) was lower than rates reported. The fact that the rate of *pneumonia did not increase over time with exposure to enfuvirtide (see Fig. 2) argues against any cumulative toxicity.
It is unclear if the increased incidence of *pneumonia reported here is related to enfuvirtide use. Potential mechanisms may include immune reconstitution; impairment of neutrophil, monocyte, or monocyte-derived dendritic cell function; or an effect on immunoglobulin A. A number of preclinical studies implemented to explore these possibilities suggest that enfuvirtide does not exert an immunosuppressive effect in several in vivo and in vitro models, however. Enfuvirtide did not impair the clearance of infectious organisms in rats subjected to challenge with influenza or S. pneumoniae.21 In vitro, enfuvirtide did not impair maturation or function of human dendritic cells and did not activate the N-formyl peptide receptor (ie, it did not suppress interleukin-12 or interleukin-10 secretion in human monocytes), nor did it have an impact on neutrophil function as measured by secretion of key cytokines in human beings and rodents.22,23
A large observational cohort study has been initiated to investigate this issue further. This chart review study plans to enroll patients starting enfuvirtide alongside matched controls (approximately 3000 patients) and to assess the onset of pneumonia over time in these 2 groups. Because of the TORO findings, however, HIV-1-infected patients receiving enfuvirtide should be carefully monitored for signs and symptoms of pneumonia, especially if they have underlying conditions that may predispose them to pneumonia or have other risk factors for pneumonia such as a low baseline CD4 cell count, a high baseline viral load, intravenous drug use, tobacco use, or a history of lung disease.
CONCLUSION
Overall, this analysis of pooled safety data from the pivotal phase 3 TORO trials indicates that enfuvirtide is a well-tolerated antiretroviral agent with a maintained safety profile over 48 weeks in treatment-experienced HIV-1-infected adults. Despite high rates of ISRs and lymphadenopathy attributable to its route of administration, few patients discontinued enfuvirtide because of difficulty with injecting or as a result of ISRs or lymphadenopathy. The addition of enfuvirtide to an optimized background antiretroviral regimen did not exacerbate toxicities associated with antiretroviral therapy, and patients treated with enfuvirtide experienced a significantly lower incidence of diarrhea and other gastrointestinal side effects compared with patients in the control group. Rates of other AEs favored enfuvirtide or were generally comparable between treatments.
Background
In March 2003, enfuvirtide (T-20 or FUZEON; Roche, Basel, Switzerland), the first agent belonging to a new mechanistic class of antiretroviral drugs, the fusion inhibitors, was approved by the US Food and Drug Administration for the treatment of HIV-1 infection. Approvals in the European Union and other countries have followed since that date. Enfuvirtide interferes with the gp41-mediated fusion of the viral envelope to the host cell membrane, thus blocking the final step in viral entry to the host cell. Enfuvirtide is formulated for parenteral self-administration by the subcutaneous route, with an adult dose of 90 mg administered twice daily. This mode of administration is unique in current antiretroviral therapy, but data from clinical trials suggest that most patients find it tolerable and that it does not have a great impact on their general activities of daily living.1
Tolerability of antiretroviral agents is a critical factor contributing to treatment outcome, with medication side effects significantly influencing patient adherence to highly active antiretroviral therapy (HAART)2 and an estimated 1 in 4 patients discontinuing therapy within 12 months of initiation because of drug toxicity.3 In particular, gastrointestinal adverse events (AEs) have been identified as the most frequently cited reason for discontinuation of HAART regimens among HIV-infected individuals.4
There is thus a growing need for new, potent, and well-tolerated antiretroviral agents, particularly for treatment-experienced patients who harbor viral strains that have lost susceptibility to conventional agents. Enfuvirtide in combination with an optimized background of oral antiretroviral drugs demonstrates potent activity against HIV-1 strains that are resistant to all 3 of the other available antiretroviral drug classes and has demonstrated durable efficacy in heavily treatment-experienced patients across a range of clinical trials for up to 96 weeks of treatment.5 The safety and efficacy of enfuvirtide in treatment-experienced HIV-1-infected individuals was assessed in 2 pivotal phase 3 trials: T-20 Versus Optimized Background Regimen Only (TORO) 1, conducted in the United State, Canada, Mexico, and Brazil, and TORO 2, conducted in Europe and Australia.6,7 The pooled 24-week analyses of these open-label, randomized, controlled trials showed that enfuvirtide added to an optimized background regimen of antiretroviral agents has a favorable safety profile and significantly improves immunologic and virologic responses compared with an optimized background regimen alone.8 Results from the prospectively planned week 48 analysis of the pooled TORO database have demonstrated the durable benefits of enfuvirtide treatment.8a Here, the pooled 48-week safety data from the TORO 1 and TORO 2 studies are presented.
RESULTS
Baseline Parameters and Follow-Up
Of the 1013 patients randomized in the 2 TORO studies, 997 received at least 1 dose of study medication with at least 1 postbaseline assessment and were included in the pooled 48-week safety population: 663 patients in the enfuvirtide group and 334 patients in the control group. The median baseline viral loads of these patients were 5.2 and 5.1 log10 copies/mL for patients randomized to the enfuvirtide and control groups, respectively, and the median baseline CD4 cell counts were 88 and 97 cells/mm3, respectively. Patients had received a median of 12 prior antiretroviral agents, and the 2 treatment groups had similar experience of antiretroviral agents in terms of the number and duration of previous nucleoside analogues, nonnucleoside analogues, and protease inhibitors. More than 80% of TORO patients had experienced an AIDS-defining event before the study. Other baseline characteristics of the study population were similar and are detailed in previous reports.6,7
Withdrawals
Patient disposition at week 48 and reasons for withdrawal are summarized in Table 1. With the exception of ISRs, in neither treatment group was a specific AE responsible for >1% of patients discontinuing. Few patients (<1%) discontinued because of difficulty with enfuvirtide administration.
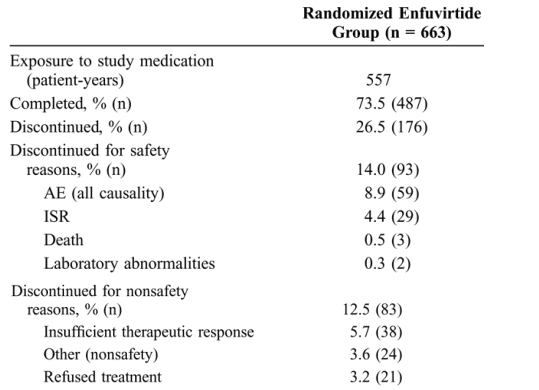
Local injection Site Reactions
Local ISRs were the most common AE associated with enfuvirtide treatment, with 98.3% of enfuvirtide-treated patients in these studies experiencing an ISR. Most patients (73.8%) had their first ISR during week 1 of enfuvirtide treatment, with overall grading based on pain or discomfort (Fig. 1). The proportion of patients with mild, moderate, or severe pain or discomfort remained steady during the treatment period, with 46% to 53% having mild tenderness, 15.1% to 19.7% having moderate pain, and 0.9% to 3.3% reporting severe pain requiring analgesics or limiting usual activities at any given visit (see Fig. 1). Most patients (71.1%) had between 1 and 5 reactions evident at a study visit, and the average duration of an individual ISR was ≦3 days for 34% of patients, >3 days and ≦7 days for 42% of patients, and >7 days for 24% of patients.
ISRs were rarely treatment limiting, as evidenced by the small number of enfuvirtide treatment discontinuations because of a reaction (4.4% in the randomized enfuvirtide group and 4.5% in the switch patient group). For most patients, these reactions were associated with mild tenderness or moderate pain at the injection site. Cumulatively, over the 48-week treatment period, 11% of patients had severe pain (grade 3) at some point during the study that required prescription of nontopical analgesics or limited their usual activities; no patient, however, had severe pain that met the criteria for an SAE (grade 4). The most frequent signs and symptoms of a local ISR were pain and/or discomfort (96.1%), erythema (90.8%), induration (90.2%), and nodules and cysts (80.4%). There was no evidence of an increase in severity over time for any of the signs and symptoms of a local ISR (see Fig. 1), and few patients (1.8 events per 100 patient-years in the enfuvirtide group, 5.0 events per 100 patient-years in the switch patient group, and 2.4 events per 100 patient-years in the combined enfuvirtide group) experienced infections at the injection site.
Of those patients with erythema, 23.8% had grade 3 (worst average grade) erythema (≥50 mm and <85 mm) and 10.5% had grade 4 erythema (≥85 mm), whereas among patients with induration, 43.5% experienced grade 3 induration (≥23 mm and <50 mm) and 19.4% experienced grade 4 induration (>50 mm). Of those patients with nodules and cysts, 29.1% had lesions >30 mm (grade 3). One patient (0.2%) had grade 4 nodules and cysts associated with grade 4 erythema and ecchymosis and grade 2 overall pain and discomfort at week 48.
A trend toward more severe ISRs was observed in patients with lower CD4 cell counts, a lower body mass index, and no treatment-emergent eosinophilia. There were no other apparent trends for the severity and frequency of the ISRs in any of the other subgroup categories examined, including age, gender, race, baseline CD4 cell counts, eosinophilia, and fat redistribution.
Lymphadenopathy
Lymphadenopathy (all causality) occurred at an incidence of 5.9, 12.5, and 7.1 events per 100 patient-years in the randomized enfuvirtide, switch patient, and combined enfuvirtide groups, respectively, compared with 1.2 events per 100 patient-years in the control group. The differences in the rates of lymphadenopathy between the combined enfuvirtide group and the control group were significant (P = 0.015). Of the 7.1 events per 100 patient-years of lymphadenopathy in the combined enfuvirtide group, 2.2 events per 100 patient-years were considered by the investigator to be treatment related. No events of lymphadenopathy were severe or life threatening and no patients withdrew from the studies because of this event.
Hypersensitivity Reactions to Enfuvirtide
Six cases (<1%) of systemic hypersensitivity reaction that were considered to be related to enfuvirtide were observed during the 48-week period, and in 5 cases, it recurred on rechallenge. Signs and symptoms included (individually and in combination) rash, fever, nausea and vomiting, chills, rigors, hypotension, and elevated serum liver transaminases.
*Pneumonia (collapsed term)
*Pneumonia (all causality), which was observed in 45 subjects (6.7 per 100 patient-years) in the combined enfuvirtide group and in 1 subject (0.6 per 100 patient-years) in the control group (Table 3), was the only collapsed term for which the difference between the treatment groups was statistically significant. Of the 37 cases of *pneumonia in the randomized enfuvirtide group (6.6 per 100 patient-years of exposure) and the 8 cases in the switch patient group (6.7 per 100 patient-years of exposure), 7 and 2 cases, respectively, occurred within the first 30 days of treatment with enfuvirtide. A display of the number of cases per 8-week period (Fig. 2) demonstrated a slightly higher rate in the first 8 weeks followed by a fairly constant rate thereafter, with no increasing rate that might indicate cumulative or latent toxicity. Approximately half of the study subjects with *pneumonia required hospitalization. Except for the 3 deaths noted previously, all *pneumonia patients responded to antibiotics with resolution and all except 1 were able to continue in the study.
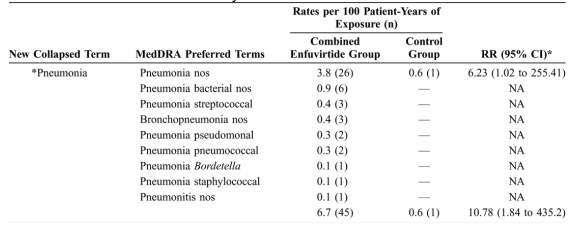
Many (64%) of the patients exposed to enfuvirtide who experienced *pneumonia during the TORO studies had a low (<50 cells/mm3) CD4 cell count at study baseline. At the time they developed *pneumonia, 62% had CD4 cell counts <200 cells/mm3, 42% had cell counts <100 cells/mm3, and 27% had cell counts <50 cells/mm3. In addition to a low baseline CD4 cell count, most *pneumonia patients had between 1 and 4 other risk factors that potentially predisposed them to developing pneumonia, including prior (33%) or current (36%) tobacco use, prior lung disease (47%), and intravenous drug abuse (13%). Most also had concurrent infections or illnesses, including upper respiratory infection, bronchitis, sinusitis, otitis, cellulitis, CMV, Kaposi sarcoma, or presumptive toxoplasmosis, during the onset of *pneumonia. For patients in whom the infective organism was isolated, these organisms were common causes of pneumonia, including Streptococcus pneumoniae, Staphylococcus aureus, and Pseudomonas. Seventy-eight percent of patients were on some type of prophylactic antibiotics for at least 7 days before the onset of *pneumonia.
Clinical Laboratory Tests TOP
Most patients had no change from baseline in the toxicity grade of any laboratory parameter. Exposure-adjusted treatment-emergent grade 3 or 4 laboratory abnormalities tended to show higher rates in the control group than in the randomized enfuvirtide and combined enfuvirtide groups, although these differences were not statistically significant (data not shown).
The overall rate of treatment-emergent eosinophilia (>0.7 X 109 cells/L, the upper limit of normal [ULN]) was higher in the randomized enfuvirtide and combined enfuvirtide groups than in the control group (12.9, 12.4, and 5.6 per 100 patient-years, respectively). The difference in the eosinophilia rate between the combined enfuvirtide arm and the control arm was significant (P = 0.021; RR = 2.24, 95% CI: 1.12 to 5.06). When using a higher (>1.4 X 109 cells/L, 2 X ULN) threshold for eosinophilia, similar rates were seen for all 3 groups (2.2, 1.8, and 1.8 per 100 patient-years, respectively). Treatment-emergent eosinophilia was not associated with clinical events of hypersensitivity or with a higher frequency of ISRs. A trend toward more severe ISRs was observed in patients who did not experience treatment-emergent eosinophilia. Review of patient records for patients with eosinophilia did not reveal any correlation with clinical symptoms suggestive of hypersensitivity to enfuvirtide.
Enfuvirtide was not associated with any adverse lipid or glycemic laboratory parameters,12 and vital signs and electrocardiograms showed no evidence of toxicity associated with enfuvirtide treatment.
Adverse Events
Overall Summary
Most patients enrolled in the TORO trials experienced at least 1 AE (all causality, excluding local ISRs) during the 48-week study period (97.1% in the randomized enfuvirtide group, 89.6% in the switch patient group, 95.3% in the combined enfuvirtide group, and 90.7% in the control group). The overall all-causality non-ISR AE rates were lower in the randomized enfuvirtide (115.6 events per 100 patient-years) and combined enfuvirtide (124.6 events per 100 patient-years) groups than in the control group (186.9 events per 100 patient-years). Most (98.3%) of enfuvirtide-treated patients in these studies had a local ISR, and these are reported separately elsewhere in this article. All-causality AEs were otherwise varied, with no specific patterns. Total exposure to study medication and a list of exposure-adjusted AEs are shown in Table 2.
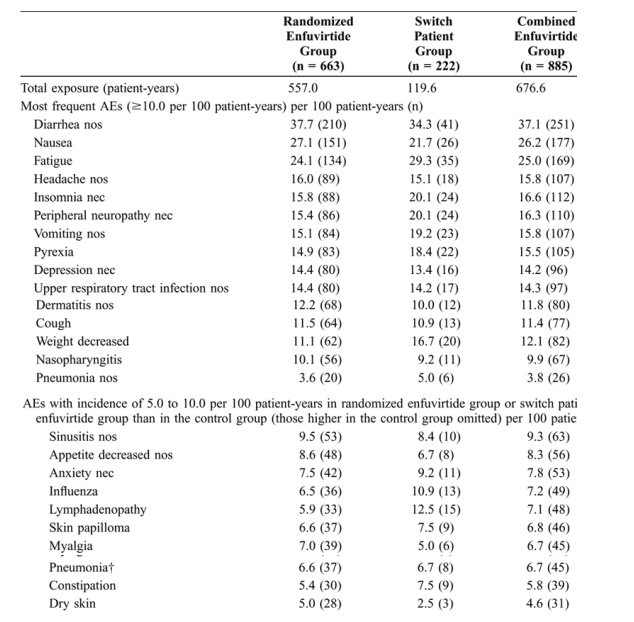
The overall number of treatment-related AE rates was lower in the randomized enfuvirtide (96.2 events per 100 patient-years) and combined enfuvirtide (100.9 events per 100 patient-years) groups than in the control group (149.9 events per 100 patient-years). Diarrhea, nausea, and fatigue were the most frequently reported all-causality AEs and treatment-related AEs for all groups, including the switch patient group, over 48 weeks, and all occurred significantly less frequently in patients taking enfuvirtide.
Pneumonia nos (relative risk [RR] = 6.23), lymphadenopathy (RR = 5.75), vertigo (RR = 5.27), and weakness (RR = 4.31) were among the AEs (all causality), with the highest RR for the combined enfuvirtide group compared with the control group. For weakness and vertigo, however, the incidence in the combined enfuvirtide group was <5 events per 100 patient-years for both (2.7 events per 100 patient-years and 3.3 events per 100 patient-years, respectively). The only all-causality AEs that were statistically significantly more frequent in the combined enfuvirtide group than in the control group were pneumonia nos and lymphadenopathy, and in both cases, the CI was wide (95% CI: 1.02 to 255.41 and 1.51 to 48.85, respectively).
For treatment-related AEs (data not shown), anxiety (RR = 5.03), lymphadenopathy (RR = 3.59), renal calculus (RR = 3.11), and weakness (RR = 3.11) were the events with the highest enfuvirtide treatment-related RRs. No treatment-related AEs were statistically significantly more frequent in the combined enfuvirtide group than in the control group, however.
Deaths and Serious Adverse Events
Overall, 16 patients (2.4%) in the randomized enfuvirtide group and 5 patients (1.0%) in the control group had an event with a final outcome of death and with an onset during treatment or within 28 days of the last dose of medication. Two patients in the switch patient group died before week 48. Adjusting for exposure, rates of deaths were similar in the randomized enfuvirtide (2.7 deaths per 100 patient-years), switch patient (1.7 deaths per 100 patient-years), combined enfuvirtide (2.7 deaths per 100 patient-years), and control (3.1 deaths per 100 patient-years) groups. Causes of death were varied.
Three deaths among subjects who received enfuvirtide were attributed to *pneumonia; however, all 3 patients had low (<50 cells/mm3) CD4 cell counts and serious concomitant AIDS-related illnesses (lymphoma, presumptive cerebral toxoplasmosis, and ongoing treatment of cytomegalovirus [CMV] and Kaposi sarcoma) that contributed to their deaths.
Two deaths were reported as related to enfuvirtide treatment. One death was a suicide in a patient with a history of depression. In the opinion of the investigator, the depression was not related to a pharmacologic effect of enfuvirtide but rather to the patient's inability to self-inject enfuvirtide. Another patient died 6 months after the last dose of enfuvirtide because of membranoproliferative glomerulonephritis, the onset of which occurred while receiving enfuvirtide and which was considered by the investigator to be related to enfuvirtide. One patient experienced Guillain-Barre syndrome, which was reported as related to enfuvirtide, but the patient's death subsequent to aspiration pneumonia was considered by the investigator as unrelated to enfuvirtide.11
The overall SAE rate (all causality) was lower in the randomized enfuvirtide and combined enfuvirtide groups than in the control group (35.5, 36.7, and 53.0 events per 100 patient-years, respectively). The most frequently reported all-causality SAEs were varied, and no pattern was identified. In the randomized enfuvirtide and combined enfuvirtide groups, the most frequently reported all-causality SAEs were increased blood creatine phosphokinase and increased ƒÁ-glutamyl transferase; however, both occurred at higher rates in the control arm. Cellulitis was reported as an all-causality SAE in 5 patients (0.7 per 100 patient-years) in the combined enfuvirtide group and in 1 patient (0.6 per 100 patient-years) in the control group (RR = 1.20; 95% CI: 0.13 to 56.67).
|
|
| |
| |
|
|
|