| |
Insulin resistance, adiponectin, cytokines in NASH: Which is the best target to treat?
|
| |
| |
Journal of Hepatology
Volume 44, Issue 2, Pages 253-261 (February 2006)
Claire Z. Larter, Geoffrey C. Farrell
The Storr Liver Unit, Westmead's Millennium Institute, University of Sydney at Westmead Hospital, Westmead, NSW 2145, Australia
Article Outline
- 1. Introduction
- 2. Insulin resistance is universal in NAFLD/NASH
- 2.1. Clinical relevance of insulin resistance in NAFLD/NASH
- 3. Adipocytokines counter steatosis and steatohepatitis
- 3.1. Clinical relevance of adipocytokines in NAFLD/NASH
- 4. Nuclear receptors and transcription factors regulate hepatic lipid synthesis and storage
- 4.1. Peroxisome proliferator-activated receptors
- 4.2. LXR, SREBP-1c and ChREBP
- 4.3. PPAR-_ coactivators (PGC)
- 4.3.1. Clinical relevance of the transcriptional regulation of hepatic lipid homeostasis
- 5. Inflammation, oxidative stress and the role of cytokines
- 5.1. Clinical implications of oxidative stress and cytokines in NAFLD/NASH
- 6. Summary and conclusions
"....Because steatosis and insulin resistance can cause or potentiate each other, it remains elusive as to whether steatosis or insulin resistance is the initiating step in NAFLD....there is also mounting evidence that steatosis can arise from other causes and then itself give rise to hepatic insulin resistance. These causes might include primary or age-related defects in mitochondrial B-oxidation [16], but enhanced fatty acid synthesis [17], or impaired secretion of TAG-rich very low density lipoproteins (VLDL) [18] could likewise initiate steatosis....
..... The practical application of this pathobiological background is that patients presenting with NAFLD should be assessed for insulin resistance and glucose intolerance. Thus, several studies have shown that diabetes is associated with a substantially higher risk of cirrhosis in NAFLD [22,23], while recent studies find that glucose intolerance is associated with both a higher prevalence of NAFLD [24] and with more rapid progression of hepatic fibrosis [25]. Accordingly, all patients suspected to have NASH should have a short 75g glucose tolerance test with fasting, 1 and 2h blood glucose and insulin levels. Hyperinsulinemia, raised c-peptide levels (which reflect the rate of insulin synthesis), and hyperglycaemia (if present) can then be useful markers for the efficacy of the therapeutic interventions discussed later.....
..... Despite these promising findings, glitazones should not yet be used routinely in patients with NASH because of the relatively high rate of adverse effects (-10%) and because weight gain is a common unwanted effect. Thus, two-thirds of patients gained weight in one study [71], and mean body mass increased by -4% in the other [72]. The long-term effects of increasing weight gain and worsening obesity, despite the temporary improvement in insulin sensitivity, is unclear.....
....Other approaches to correct insulin resistance resulting from obesity should be preferred as first line therapy for NASH, unless the patient has glucose intolerance or established diabetes. In the latter case, use of a glitazone could be indicated to improve glycaemic control as well as lipid metabolism; it might be expected that liver disease would improve also, at least temporarily....."
6. Summary and conclusions
Distinct pathways govern the regulation of hepatic lipid synthesis and disposal. There are interactions between these pathways, and it is critical that a balance between them is maintained to prevent lipotoxicity. Disturbances within the regulatory systems of lipid partitioning result in steatosis; correcting such dysregulation by dietary or pharmacological approaches should provide a sound therapeutic approach to treatment of steatosis and steatohepatitis. Any pathogenic concept for NAFLD/NASH must account for the strong links with overnutrition and underactivity, genetic factors, and peripheral and hepatic insulin resistance, but it remains unclear whether hepatic insulin resistance is primary or secondary to steatosis. How these factors interact to expand hepatic lipid stores, cause hepatocellular injury, recruit inflammation and promote fibrosis in the liver is the current focus of investigation. Lipotoxicity, oxidative stress, release of cytokines and other pro-inflammatory mediators may each play a pro-inflammatory role, while PPAR-_ (and possibly PPAR-_), adiponectin and suppression of oxidative stress (e.g. by induction of antioxidant enzymes) may be protective pathways.
Despite these complexities, the present 'gold standard' for treatment of NAFLD is weight reduction-or more precisely, a reduction in central obesity so as to reverse insulin resistance. The lesson of diabetes intervention studies as well as from open studies of NASH is that this can be achieved by combining dietary measures with increased levels of physical activity [87,88]. Such 'lifestyle adjustment' or anti-obesity measures (including bariatric surgery when required) improve insulin sensitivity with only modest (2-8kg) weight reduction. In the majority of cases this impressively reduces liver cell injury, inflammation and hepatic fibrosis, as well as steatosis [89]. The same potential for 'unwinding' fibrotic NASH is indicated by studies of the 'glitazones', PPAR-_ agonists that also correct peripheral insulin resistance. However, these agents improve liver disease at the expense of increasing subcutaneous fat mass so as to worsen obesity. The therapeutic and preventive challenges, therefore, may be to approach NASH as a public health measure linked to overnutrition and under-activity, rather than to focus on potential pharmacological solutions that may be of only temporary benefit. Mechanisms of disease can provide new insights for prevention and therapy, but the target to treat in NASH is the affected person, and how to motivate them and our societies to adopt a healthier lifestyle.
1. Introduction
In community studies, NAFLD correlates with central adiposity, obesity, insulin resistance, and the complications of insulin resistance-the metabolic syndrome and type 2 diabetes mellitus [1,2]. Studies of histologically confirmed NASH show a near universal association with insulin resistance, and about five out of six patients have the metabolic syndrome [1-3]. The metabolic factors associated with NAFLD/NASH are also determinants of disease progression in hepatitis C and alcoholic liver disease [4,5], providing further evidence that they exert important pathobiological effects on the liver. Understanding how these metabolic factors can be assessed, and how abnormalities can be reversed is crucial for better management of patients with NASH.
2. Insulin resistance is universal in NAFLD/NASH
Insulin regulates the uptake, oxidation and storage of fuel within insulin-sensitive tissues-the liver, skeletal muscle and adipose tissue. Insulin effects this energy regulation via intracellular signalling cascades that originate at the insulin receptor. As depicted in Fig. 1, binding of insulin to its receptor stimulates autophosphorylation of tyrosine residues that act as docking sites for 'downstream' signalling molecules; the latter include the janus-activated kinases (JAK) and insulin receptor substrates (IRS)-1 and IRS-2. Several disease processes alter the operation of these signalling pathways. These processes include lipotoxicity (unsequested accumulation of cytotoxic fatty acids and their products in non-adipose tissues), oxidant stress and inflammation; all may be pertinent to NASH pathogenesis. The pathobiological mechanisms for blocking insulin receptor signalling include serine/threonine phosphorylation of IRS-1 and -2 (which abrogates physiological tyrosine phosphorylation), and competition for docking sites by suppressors of cytokine signalling (SOCS), as recently reviewed in The Journal [6].
Fig. 1. Causes of hepatic insulin resistance. Obesity induces sub-acute inflammation that promotes insulin resistance by increasing hepatic SOCS expression (via IL-6). SOCS inhibits insulin signalling directly by competing for phosphorylation sites on the insulin receptor, and indirectly by inducing SREBP-1c. In turn, SREBP-1c suppresses IRS2 synthesis, and increases fatty acid synthesis. Hepatic levels of fatty acyl-CoA increase in response to serum NEFA (fatty acids arriving from peripheral lipolysis), dietary intake and endogenous fatty acid synthesis, or from impaired B-oxidation (possibly the result of mitochondrial injury). This increase in fatty acyl-CoA (and possibly other lipid metabolites) activates PKC-O to catalyse serine/threonine phosphorylation of the IRS proteins, and/or IKK-B to activate NF-kB with release of IL-6 and augmentation of SOCS induction.
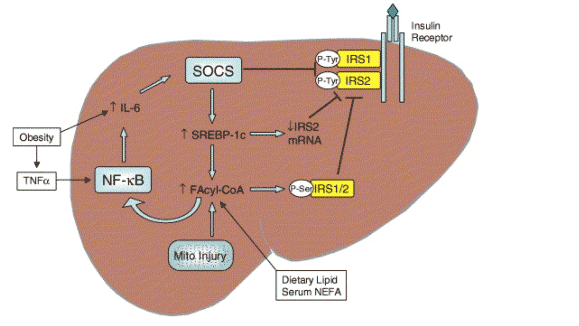
In both liver cells and myocytes, insulin resistance is strongly associated with increased cellular content of fatty acids and their metabolites (fatty acyl-CoAs, diacylglycerides and ceramides) [7,8]. The favoured mechanism by which these molecules disrupt insulin receptor signalling is activation of protein kinase C (PKC) isoforms, theta (_) in liver, epsilom (_) in muscle (rev. in [9]). In addition to PKC, other tissue kinases (c-jun N-terminal kinase (JNK) and the I_ kinase-_ (IKK)-_) also phosphorylate IRS-1 and -2 at serine/threonine sites to inhibit tyrosine phosphorylation (Fig. 1).
The physiological consequences of insulin resistance depend on whether the impairment of insulin receptor signalling is in peripheral tissues or liver. Peripheral insulin resistance impairs uptake of glucose from blood into skeletal muscle and adipose tissue; serum non-esterified fatty acid (NEFA) levels may also be elevated due to the failure of insulin to suppress lipolysis [10]. In contrast, the main consequence of hepatic insulin resistance is unrestrained hepatic glucose production resulting from impaired glycogen synthesis and failure to suppress gluconeogenesis due to impaired IRS-2 phosphorylation [10,11].
Because steatosis and insulin resistance can cause or potentiate each other, it remains elusive as to whether steatosis or insulin resistance is the initiating step in NAFLD. The older view of the relationship was that fasting hyperinsulinaemia caused by insulin resistance, in the presence of increased circulating levels of glucose and NEFA, enhances uptake of fatty acids at the same time that hepatic fatty acid synthesis is increased (see later). This traditional, 'portal theory' for steatosis resulting from insulin resistance envisages a role for visceral adiposity as the source of NEFA delivered to the liver [12]. Evidence for this has come from dogs fed a high fat diet [13]. In these animals, fat feeding expanded visceral adipose mass and promoted lipolysis, implying increased NEFA delivery to the liver, and this was associated with hepatic insulin resistance. However, although hepatic triacylglyceride (TAG) levels were elevated, peripheral blood NEFA levels remained unchanged (portal levels were not measured). Recently, it has been shown that the majority (60%) of hepatic TAG in patients with NAFLD/NASH arises from NEFA, supporting the role of peripheral lipolysis in contributing to hepatic steatosis [14]. Others have found an increase in serum NEFA levels in patients with NASH [15]. Measurements of portal NEFA are still needed to fully test the portal theory. However, there is now compelling evidence that the major source of hepatic TAG in NAFLD is the periphery.
Notwithstanding this, there is also mounting evidence that steatosis can arise from other causes and then itself give rise to hepatic insulin resistance. These causes might include primary or age-related defects in mitochondrial _-oxidation [16], but enhanced fatty acid synthesis [17], or impaired secretion of TAG-rich very low density lipoproteins (VLDL) [18] could likewise initiate steatosis (see Fig. 1). The possibility that hepatic lipids could initiate insulin resistance was first suggested by Kraegen et al. [19]. It has more recently been confirmed by Samuel et al. [20] on the basis of short-term studies of high-fat feeding in rats. After 3 days on the high-fat diet, hepatic levels of fatty acyl-CoA increased, as well as TAG. There were no such changes in peripheral tissues, and this dietary regimen failed to alter peripheral insulin sensitivity. However, hepatic insulin resistance was evident at this time as shown by impaired insulin-stimulated phosphorylation of IRS-1 and -2 and reduced capacity to activate glycogen synthesis or suppress hepatic glucose production [18,19]. In other work, liver-specific expression of lipoprotein lipase increased hepatic lipid content and caused hepatic but not muscle insulin resistance [21], while muscle-specific expression of lipoprotein lipase had the opposite effects. In addition to these effects of lipid metabolites on insulin receptor signalling, oxidative stress and inflammation can cause insulin resistance, as discussed later.
2.1. Clinical relevance of insulin resistance in NAFLD/NASH
The practical application of this pathobiological background is that patients presenting with NAFLD should be assessed for insulin resistance and glucose intolerance. Thus, several studies have shown that diabetes is associated with a substantially higher risk of cirrhosis in NAFLD [22,23], while recent studies find that glucose intolerance is associated with both a higher prevalence of NAFLD [24] and with more rapid progression of hepatic fibrosis [25]. Accordingly, all patients suspected to have NASH should have a short 75g glucose tolerance test with fasting, 1 and 2h blood glucose and insulin levels. Hyperinsulinemia, raised c-peptide levels (which reflect the rate of insulin synthesis), and hyperglycaemia (if present) can then be useful markers for the efficacy of the therapeutic interventions discussed later.
3. Adipocytokines counter steatosis and steatohepatitis
The detrimental effects of systemic insulin resistance on hepatic lipid partitioning are opposed by adipocytokines, humoral mediators arising from the adipose tissue. In the lipodystrophies, subcutaneous fat atrophy is associated with a virtual absence of circulating leptin. In these syndromes, as well as in mice with congenital lipodystrophy, leptin injection reverses fatty liver disease [26,27]. However, in cases of NASH associated with obesity, insulin resistance and the metabolic syndrome, serum leptin levels are increased in proportion to the severity of steatosis [28-30]. It has therefore been proposed that the liver becomes refractory to the 'anti-steatotic' effects of leptin, a state of 'hepatic leptin resistance' that accompanies hepatic insulin resistance rather than correcting it [28]. Leptin infusion is therefore unlikely to be of therapeutic benefit for most patients with NAFLD/NASH.
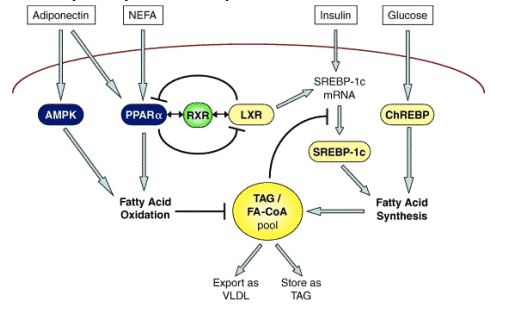
Adiponectin is more closely implicated in the pathogenesis of NAFLD/NASH. Thus, serum adiponectin levels are inversely related to hepatic fat stores [31,32], and in one study serum adiponectin values were lower in NASH patients than in persons with uncomplicated steatosis [32]. In another, hepatic expression of adiponectin and its type II receptor were less in NASH than in fatty liver [33]. The actions of adiponectin on the liver are to oppose fatty acid synthesis, and promote mitochondrial _-oxidation; as depicted in Fig. 2, these actions are exerted through activation of the cyclic-AMP dependent protein kinase (AMPK) [34]. Adiponectin also exerts anti-inflammatory effects by opposing the synthesis and release of TNF-_ from macrophages within adipose tissue in obesity [35]. It is therefore of great interest that administration of exogenous adiponectin reverses experimental forms of NAFLD and steatohepatitis [36]. It has also been suggested that adiponectin could exert some of its hepatic lipid modulatory and anti-inflammatory effects via peroxisome proliferator-activated receptor-_ (PPAR-_) [36], pharmacological activation of which completely reverses experimental steatohepatitis [37]. More indirectly, PPAR-_ agonists, such as the thiazolidinediones (discussed later) stimulate adiponectin synthesis [38], which could then act via PPAR-_ co-activator-1_ (PGC) to activate PPAR-_ [39].
Fig. 2. Hepatic lipid homeostasis. Lipid homeostasis is achieved by balancing fatty acid synthesis with fatty acid oxidation and export. Fatty acid synthesis is induced by insulin and glucose via the transcription factors SREBP-1c and ChREBP, respectively. LXR also promotes fatty acid synthesis by inducing SREBP-1c. Fatty acid oxidation is promoted by PPAR-a and the protein kinase, AMPK. Adiponectin, an adipocytokine, increases hepatic fatty acid oxidation by inducing PPAR-a and AMPK; increased levels of hepatic fatty acids also induce PPAR-a. PPAR-a and LXR have opposite functions but their engagement of the same heteropartner, RXR, enables hepatocytes to transiently commit to either fatty acid synthesis or fatty acid oxidation.
3.1. Clinical relevance of adipocytokines in NAFLD/NASH
While serum adiponectin levels can be detected with commercially available ELISA assays [27], it is not yet clear whether a single fasting adiponectin value can reliably distinguish an individual patient with NASH from someone with only steatosis. Inclusion of serum adiponectin in predictive indices for non-invasive assessment of patients with NAFLD is an exciting future development in clinical care. Adiponectin replacement therapy in not yet available as a treatment option, but factors which stimulate adiponectin production, such as dietary regimens and PPAR-_ agonists are likely to be promising in the future. Likewise, PPAR-_ agonists, which include fibrates have been shown to improve liver tests in patients with NASH [40,41]. More prolonged trials with carefully designed histological endpoints may be of value, provided safety is not a dose-limiting issue.
4. Nuclear receptors and transcription factors regulate hepatic lipid synthesis and storage
Bodily lipid homeostasis requires interactions between the liver, muscle and adipose tissue. These interactions are predominantly regulated by nuclear receptors such as the PPAR family, and liver X receptor (LXR) that operates through key transcription factors such as sterol regulatory element binding protein-1c (SREBP-1c) and carbohydrate response element binding protein (ChREBP). The role of PGCs in liver is less clear; they interact with both the PPAR family and LXR [42,43]. The primary goal of lipid homeostasis is to prevent lipotoxicity, a condition that arises from inappropriate lipid partitioning into non-adipose tissues. To achieve lipid homeostasis, it is critical to balance uptake, synthesis (lipogenesis), esterification and storage, with oxidation and export of fatty acids. In obesity-related steatosis, hepatic lipogenesis is up-regulated even though hepatic uptake of NEFA is increased.
4.1. Peroxisome proliferator-activated receptors
PPARs, the largest family of nuclear receptors, are now a prime focus of NAFLD/NASH research. There are three subtypes, PPAR-_, -_ and -_ (previously named PPAR-_). Upon receptor activation, all bind the retinoid X receptor (RXR) to form transcriptionally active heterodimers. PPAR-_ is primarily expressed in tissues that use fatty acids as a fuel, such as the liver, muscle, heart and kidneys. In contrast, PPAR-_ is found predominantly in adipose tissue where it mediates differentiation of pre-adipocytes (adipogenesis), lipid storage and insulin action. The ability of PPARs to 'sense' fatty acids is crucial for their function as transcription factors. They control multiple genes concerned with lipid homeostasis. In general, PPARs favour fatty acid catabolism in non-adipose tissues and promote fatty acid storage (as TAG) in adipose tissue; these effects enable appropriate fuel partitioning and lipid storage within the body (reviewed in [44]).
PPAR-_ is central to hepatic lipid homeostasis. PPAR-_ promotes fatty acid oxidation in muscle and will not be discussed further here. When hepatic fatty acid levels increase, PPAR-_ is activated, leading to transcription of such genes as liver fatty acid binding protein (LFABP), acyl-CoA oxidase (ACO), cytochrome P450 (CYP)4A, microsomal triglyceride transfer protein (MTTP) and apolipoprotein B100 (apoB100) [45-49]. The net effect is catabolism and clearance of fatty acids. Fatty acids may also be esterified into TAG or phospholipids for storage within hepatocytes, or for export. MTTP lipidates apoB100 to enable lipid export from the liver as VLDL. It has recently been appreciated that the liver may respond to newly synthesized fatty acids differently from those 'recycled' from peripheral stores, and this discrimination is mediated by selective effects on PPAR-_ [50]. This may explain why PPAR-_ activation does not appear to occur as an adaptive response in fatty liver disease, when it would be an effective pathway to enhance insulin sensitivity and suppress inflammatory recruitment. It should be noted, however, that the role of PPAR-_ in regulating hepatic lipid metabolism has been studied predominantly in rodents; there is evidence that PPAR-_ activation does not increase the expression of enzymes involved in peroxisomal _-oxidation to the same extent in human liver [51,52]. Thus, while pharmacologic PPAR-_ activation effectively 'cured' fibrosing steatohepatitis in a murine, dietary model [37], the efficacy of such agents is less clear in humans [40,41].
PPAR-_ is found predominantly in adipocytes. It has both opposite and complementary functions to PPAR-_. Thus, PPAR-_ activation leads to differentiation of adipocytes from pre-adipocytes [53]. This increases the lipid storage capacity of the adipose mass, and also increases the number of small, insulin-sensitive adipocytes so as to improve insulin sensitivity [54]. Increasingly, the importance of PPAR-_ is being recognized in the liver, despite the relatively low levels of expression normally found there. Steatosis is often associated with hepatocyte expression of PPAR-_. In some situations, such PPAR-_ expression precedes and in others it appears secondary to lipid accumulation [55,56]. It is currently unclear whether the resultant adipocytic transformation of hepatocytes (adipogenesis) functions to accommodate TAG storage, thereby conferring protection from lipotoxicity to lipid-laden hepatocytes, or whether it is a part of the hepatocellular proliferative response to injury [57].
4.2. LXR, SREBP-1c and ChREBP
Originally described as a nuclear receptor that regulates cholesterol homeostasis, LXR is now also known to mediate lipid homeostasis. Like the PPARs, ligand binding stimulates LXR to heterodimerize with RXR and form transcriptional transactivation complexes. LXR contributes to the regulation of SREBP-1c [58], which with ChREBP regulates endogenous lipid synthesis in the liver [59]. SREBP-1c and ChREBP activate similar lipogenic genes, such as fatty acid synthase (FAS) and acetyl CoA carboxylase [59-61]. Lipogenesis is generally induced by insulin-mediated induction of SREBP-1c, but can also be activated by glucose via ChREBP [62]. In the liver, LXR-induced activation of SREBP-1c promotes fatty acid synthesis and may therefore induce steatosis. Agonist induction of LXR causes severe steatosis when diabetes is present [63]. The role of LXR-dependent induction of SREBP-1c may be different in muscle [64], while in adipose tissue LXR initiates adipocyte differentiation and lipogenesis through induction of PPAR-_ [65].
LXR interacts with PPAR-_ in a reciprocally inhibitory manner, so that the two factors can exert their opposite functions on lipid metabolism. Thus, as depicted in Fig. 2, LXR promotes lipid synthesis while PPAR-_ promotes lipid disposal. Competition for the RXR allows activation of one nuclear receptor to sequester available RXR, thereby preventing activation of the other receptor [66,67]. This exclusive mechanism of regulation enables transient commitment of cells to either lipid synthesis or lipid disposal, ensuring rapid and effective adjustments to lipid balance (Fig. 2).
Under physiological conditions, SREBP-1c is transiently induced in the liver by insulin through activation of IRS-2; this causes a switch from glycogen synthesis to lipid synthesis. To complete a feedback loop, SREBP-1c then suppresses IRS-2 transcription. Under certain pathogenic conditions, expression of SREBP-1c in the liver remains elevated, and this increases lipid synthesis with resultant accumulation of fat. At the same time, the associated decrease in IRS-2 expression induces or exacerbates hepatic insulin resistance (see Fig. 1) [68].
4.3. PPAR-_ coactivators (PGC)
The PPAR-_ coactivator PGC-1_ is involved in the fasting response, during which it increases mitochondrial biogenesis and fatty acid _-oxidation, as well as promoting hepatic gluconeogenesis [69,70]. Activation of PGC-1_ may protect against lipid-induced insulin resistance by promoting mitochondrial _-oxidation, but it may also exacerbate hyperglycaemia by increasing hepatic glucose production.
4.3.1. Clinical relevance of the transcriptional regulation of hepatic lipid homeostasis
The potential value of PPAR-_ agonists has been mentioned, but these should not yet be used routinely. LXR agonists oppose the beneficial effects of PPAR-_ stimulation and may worsen steatosis. PPAR-_ agonist thiazolidinedione ('glitazones') are among the most promising pharmacological agents trialled against NASH [71,72]. In affected patients they improve insulin sensitivity, correct liver test abnormalities and improve all aspects of liver pathology, including hepatocellular injury, lobular inflammation and fibrosis as well as steatosis [71,72]. Despite these promising findings, glitazones should not yet be used routinely in patients with NASH because of the relatively high rate of adverse effects (-10%) and because weight gain is a common unwanted effect. Thus, two-thirds of patients gained weight in one study [71], and mean body mass increased by -4% in the other [72]. The long-term effects of increasing weight gain and worsening obesity, despite the temporary improvement in insulin sensitivity, is unclear.
Other approaches to correct insulin resistance resulting from obesity should be preferred as first line therapy for NASH, unless the patient has glucose intolerance or established diabetes. In the latter case, use of a glitazone could be indicated to improve glycaemic control as well as lipid metabolism; it might be expected that liver disease would improve also, at least temporarily.
5. Inflammation, oxidative stress and the role of cytokines
The processes involved with inflammatory recruitment in NASH can also impede insulin receptor signalling. These processes include oxidative stress and/or mobilisation of tumor necrosis factor-_ (TNF-_) and related cytokines, such as interleukin-6 (IL-6). In both NASH and experimental steatohepatitis, hepatic expression of CYP2E1 is increased leading to oxidative stress [73]. Such CYP2E1 over-expression impairs insulin receptor signalling in cultured hepatocytes as well as in experimental steatohepatitis [74]. The mechanism has been attributed to enhanced serine/threonine phosphorylation of IRS-1 and IRS-2 with correspondingly impaired tyrosine phosphorylation. IKK-_ and/or JNK could be the pathways involved. Oxidative stress has been suggested to cause IKK-_-dependent NF-_B activation in experimental steatohepatitis [75], but this remains unproven.
Circulating TNF-_ levels and hepatic expression of its type 1 receptor are increased in NASH [32,76], but do not appear to discriminate steatohepatitis from steatosis. In ob/ob mice (which develop steatosis without NASH), administration of metformin improved insulin sensitivity and liver disease by a proposed mechanism that involved TNF-_-mediated suppression of fatty acid synthesis [77]. However, others found that TNF-_ receptor signaling is not relevant in this experimental form of fatty liver disease because the phenotype of ob/ob mice was the same whether or not they were crossed with TNF-_ type 1 receptor deleted mice [78]. Diehl and colleagues have also reported that macrobiotics improved liver disease in ob/ob mice by a suggested mechanism that links the gut flora to endogenous TNF-_ production [79,80]. Conversely, nutritional steatohepatitis can be produced experimentally in both TNF-_ gene-deleted and TNF-_ type 1 receptor nullizygous mice [75,81], showing that TNF-_ is not an essential mediator of this pathology.
A potential pathogenic role of inflammation in the development of hepatic insulin resistance has been recently demonstrated [82]. Earlier studies had shown that steatosis is associated with activation of NF-_B in the liver. In order to delineate the effects of inflammation from those of steatosis, a mouse was generated with liver-specific expression of IKK-_ to cause modest, constitutive activation of hepatic NF-_B. In this model, both hepatic and peripheral insulin resistance were the consequence of NF-_B-dependent induction of IL-6 (Fig. 2). The results of this study provide support for how hepatic steatosis can precipitate both hepatic and peripheral insulin resistance through the induction of sub-acute inflammation. One likely mechanism involves the SOCS family of proteins, as recently reviewed in the Journal [6]. In the liver, SOCS-1 is a specific inhibitor of IRS-2, while SOCS-3 inhibits both IRS-1 and -2 by interacting with the insulin receptor at Tyr960 [83]. SOCS-1 and SOCS-3 can also inhibit insulin receptor signalling by ubiquitinating IRS-1 and IRS-2, targeting them for proteasomal degradation [84], or by impairing JAK-STAT-3 activation [6].
Whatever the pathogenic mechanisms by which SOCS proteins impair insulin receptor signalling pathways, it is now clear that over-expression of SOCS proteins in the liver induces hepatic insulin resistance [85]. It is also of interest that over-expression of SOCS-1 and SOCS-3 promotes SREBP-1c expression and subsequent development of steatosis and metabolic syndrome in mice [85]. The latter effects may be explained by removal of SREBP-1c from STAT-3-mediated suppression. Similarly, when SOCS-1 and/or SOCS-3 levels were normalized in obese (db/db) diabetic mice, there were improvements in both insulin signalling and steatosis. When both SOCS proteins were normalized, hepatic SREBP-1c mRNA (which is normally elevated in db/db mice) returned to wild-type levels, and steatosis and metabolic syndrome improved [85]. These observations implicate SOCS proteins as a critical link between hepatic inflammation, steatosis and insulin resistance in NASH (Fig. 1).
5.1. Clinical implications of oxidative stress and cytokines in NAFLD/NASH
The weight of evidence is now against TNF-_ as a pathogenic mechanism in experimental NAFLD/NASH, although further longitudinal studies are indicated in humans with the condition. Thus, there is not yet any role for anti-TNF-_ strategies (other than weight reduction) in the clinical management of patients with NASH. Strategies to block oxidative stress are of greater interest, with some evidence that ALT normalization or histological improvement occurs with vitamin E (alone or with vitamin C or pioglitazone) and betaine [86]. However, more definitive studies are needed before these or other antioxidants and antifibrotic agents (including silymarin) can be routinely recommended.
6. Summary and conclusions
Distinct pathways govern the regulation of hepatic lipid synthesis and disposal. There are interactions between these pathways, and it is critical that a balance between them is maintained to prevent lipotoxicity. Disturbances within the regulatory systems of lipid partitioning result in steatosis; correcting such dysregulation by dietary or pharmacological approaches should provide a sound therapeutic approach to treatment of steatosis and steatohepatitis. Any pathogenic concept for NAFLD/NASH must account for the strong links with overnutrition and underactivity, genetic factors, and peripheral and hepatic insulin resistance, but it remains unclear whether hepatic insulin resistance is primary or secondary to steatosis. How these factors interact to expand hepatic lipid stores, cause hepatocellular injury, recruit inflammation and promote fibrosis in the liver is the current focus of investigation. Lipotoxicity, oxidative stress, release of cytokines and other pro-inflammatory mediators may each play a pro-inflammatory role, while PPAR-_ (and possibly PPAR-_), adiponectin and suppression of oxidative stress (e.g. by induction of antioxidant enzymes) may be protective pathways.
Despite these complexities, the present 'gold standard' for treatment of NAFLD is weight reduction-or more precisely, a reduction in central obesity so as to reverse insulin resistance. The lesson of diabetes intervention studies as well as from open studies of NASH is that this can be achieved by combining dietary measures with increased levels of physical activity [87,88]. Such 'lifestyle adjustment' or anti-obesity measures (including bariatric surgery when required) improve insulin sensitivity with only modest (2-8kg) weight reduction. In the majority of cases this impressively reduces liver cell injury, inflammation and hepatic fibrosis, as well as steatosis [89]. The same potential for 'unwinding' fibrotic NASH is indicated by studies of the 'glitazones', PPAR-_ agonists that also correct peripheral insulin resistance. However, these agents improve liver disease at the expense of increasing subcutaneous fat mass so as to worsen obesity. The therapeutic and preventive challenges, therefore, may be to approach NASH as a public health measure linked to overnutrition and under-activity, rather than to focus on potential pharmacological solutions that may be of only temporary benefit. Mechanisms of disease can provide new insights for prevention and therapy, but the target to treat in NASH is the affected person, and how to motivate them and our societies to adopt a healthier lifestyle.
|
|
| |
| |
|
|
|