| |
The KLEAN study of fosamprenavir-ritonavir versus lopinavir-ritonavir, each in combination with abacavir-lamivudine, for initial treatment of HIV infection over 48 weeks: a randomised non-inferiority trial
|
| |
| |
The Lancet August 5 2006; 368:476-482
Prof Joseph Eron Jr MD a , Prof Patrick Yeni MD b, Joseph Gathe Jr MD c, Vicente Estrada MD d, Edwin DeJesus MD e, Prof Schlomo Staszewski MD f, Philip Lackey MD g, Prof Christine Katlama MD h, Benjamin Young MD i, Linda Yau PhD j, Denise Sutherland-Phillips MD j, Paul Wannamaker BA j, Cindy Vavro BA j, Lisa Patel PharmD j, Jane Yeo PhD k and Mark Shaefer PharmD j, for the KLEAN study team
Summary
Background
Lopinavir-ritonavir is a preferred protease inhibitor co-formulation for initial HIV-1 treatment. Fosamprenavir-ritonavir has shown similar efficacy and safety to lopinavir-ritonavir when each is combined with two nucleoside reverse transcriptase inhibitors. We compared the two treatments directly in antiretroviral-naive patients.
Methods
This open-label, non-inferiority study included 878 antiretroviral-naive, HIV-1-infected patients randomised to receive either fosamprenavir-ritonavir 700 mg/100 mg twice daily or lopinavir-ritonavir 400 mg/100 mg twice daily, each with the co-formulation of abacavir-lamivudine 600 mg/300 mg once daily. Primary endpoints were proportion of patients achieving HIV-1 RNA less than 400 copies per mL at week 48 and treatment discontinuations because of an adverse event. The intent-to-treat analysis included all patients exposed to at least one dose of randomised study medication. This study is registered with ClinicalTrials.gov, number NCT00085943.
Findings
At week 48, non-inferiority of fosamprenavir-ritonavir to lopinavir-ritonavir (95% CI around the treatment difference -4-84 to 7-05) was shown, with 315 of 434 (73%) patients in the fosamprenavir-ritonavir group and 317 of 444 (71%) in the lopinavir-ritonavir group achieving HIV-1 RNA less than 400 copies per mL. Treatment discontinuations due to an adverse event were few and occurred with similar frequency in the two treatment groups (fosamprenavir-ritonavir 53, 12%; lopinavir-ritonavir 43, 10%). Diarrhoea, nausea, and abacavir hypersensitivity were the most frequent drug-related grade 2-4 adverse events. Treatment-emergent drug resistance was rare; no patient had virus that developed reduced susceptibility to fosamprenavir-ritonavir or lopinavir-ritonavir.
Interpretation
Fosamprenavir-ritonavir twice daily in treatment-naive patients provides similar antiviral efficacy, safety, tolerability, and emergence of resistance as lopinavir-ritonavir, each in combination with abacavir-lamivudine.
Introduction
Triple combination highly active antiretroviral therapy (HAART) containing a protease inhibitor with two nucleoside reverse transcriptase inhibitors (NRTIs) has resulted in striking decreases in HIV-1-related morbidity and mortality, and is currently a standard of care regimen for initial treatment of patients infected with HIV-1.1-4 Most current antiretroviral treatment guidelines recommend lopinavir-ritonavir as a preferred protease inhibitor co-formulation for initial treatment of antiretroviral-naive patients.3,4 This recommendation was based in part on the findings of a double-blind, randomised, multicentre clinical trial (study M98-863)5 that showed superiority of lopinavir-ritonavir 400 mg/100 mg twice daily to nelfinavir 750 mg three times daily in 653 treatment-naive patients. At week 48, the proportion of patients with HIV-1 RNA of less than 400 copies per mL for lopinavir-ritonavir was 75%, versus 63% for nelfinavir (p<0-001); proportions with less than 50 copies per mL were 67% versus 52%, respectively (intent-to-treat [ITT], missing or discontinuation=failure analysis). No primary mutations associated with protease inhibitors were detected in the lopinavir-ritonavir group through 48 weeks.5
The safety and efficacy of fosamprenavir-ritonavir 1400 mg/200 mg once daily has also been compared with that of nelfinavir 1250 mg once daily in a phase III (Note from Jules: this must be a misprint, as NFV is a bid drug), randomised trial in 649 antiretroviral-naive patients (APV30002, SOLO).6 At week 48, the fosamprenavir-ritonavir regimen proved non-inferior to the nelfinavir regimen with respect to the proportion of patients achieving HIV-1 RNA less than 400 copies per mL (69% vs 68%) and less than 50 copies per mL (55% vs 53%), respectively (ITT, rebound or discontinuation=failure analysis). Drug-related diarrhoea occurred less often in the fosamprenavir-ritonavir group than in the nelfinavir group (9% vs 16%, p=0-008). No treatment-emergent primary protease-inhibitor mutations or phenotypic resistance were detected in the fosamprenavir-ritonavir group through 48 weeks. By contrast, genotypic resistance to protease inhibitors emerged in patients randomised to receive nelfinavir.7
No large, randomised clinical trials have been undertaken to date comparing two ritonavir-enhanced protease inhibitors such as lopinavir-ritonavir and fosamprenavir-ritonavir in antiretroviral-naive patients. At study initiation, lopinavir-ritonavir was approved by the US Food and Drug Administration (FDA) for twice-daily dosing and fosamprenavir-ritonavir was FDA-approved for either once or twice-daily dosing. To maintain consistency in dosing regimens and to derive clinical data with twice-daily fosamprenavir-ritonavir, we chose twice-daily dosing regimens for both protease inhibitors. We therefore undertook KLEAN (Kaletra versus Lexiva with Epivir and Abacavir in ART-Naive patients), an international, multicentre study to compare the efficacy and safety of fosamprenavir-ritonavir 700 mg/100 mg twice daily with that of lopinavir-ritonavir 400 mg/100 mg twice daily, each in combination with the abacavir-lamivudine 600 mg/300 mg fixed-dose combination tablet once daily.
Results
During the recruitment period (May, 2004, to January, 2005), 887 patients from 131 centres were randomised (figure 1). In total, 679 (77%) of the 878 patients in the ITT-E population completed week 48. The per-protocol population included 838 patients (fosamprenavir-ritonavir: 414; lopinavir-ritonavir: 424).
Baseline characteristics were similar between the two treatment groups (table 1). The ITT-E population had high median baseline HIV-1 RNA values (5-1 log10 copies per mL) and low median CD4 counts (192 cells per μL). Notably, 54% of the population had HIV-1 RNA of 100 000 copies per mL or greater and 17% had a CD4 count less than 50 cells per μL at baseline.
At week 48, the response rates for the proportion of patients who achieved HIV-1 RNA less than 400 copies per mL were 73% in the fosamprenavir-ritonavir group and 71% in the lopinavir-ritonavir group by ITT-E, TLOVR analysis (95% CI for treatment difference -4-84 to 7-05), thereby establishing non-inferiority of fosamprenavir-ritonavir to lopinavir-ritonavir (table 2, figure 2). The observed analysis of the ITT-E population showed consistent results: response rates were 96% (314 of 328) in patients receiving fosamprenavir-ritonavir and 97% (329 of 341) in patients receiving lopinavir-ritonavir at week 48 for HIV-1 RNA less than 400 copies per mL. Similar results were seen when using the more stringent endpoint of HIV-1 RNA less than 50 copies per mL at week 48: the ITT-E, TLOVR response rates were 66% (285 of 434) in the fosamprenavir-ritonavir group and 65% (288 of 444) in the lopinavir-ritonavir group (figure 2) and the ITT-E, observed rates were 89% (292 of 328) and 88% (299 of 341) in the fosamprenavir-ritonavir and lopinavir-ritonavir groups, respectively. Additional sensitivity analyses of the primary and less than 50 copies/mL endpoints indicated that the primary efficacy results were robust regardless of the analysis method used, and the two treatment groups would have been declared non-inferior using any of these methods of analysis, including: missing/discontinuation=failure, 72% versus 74% (<400 copies per mL) and 67% versus 67% (<50 copies/mL), respectively; and per-protocol observed, 96% versus 96% and 89% versus 88%, respectively.
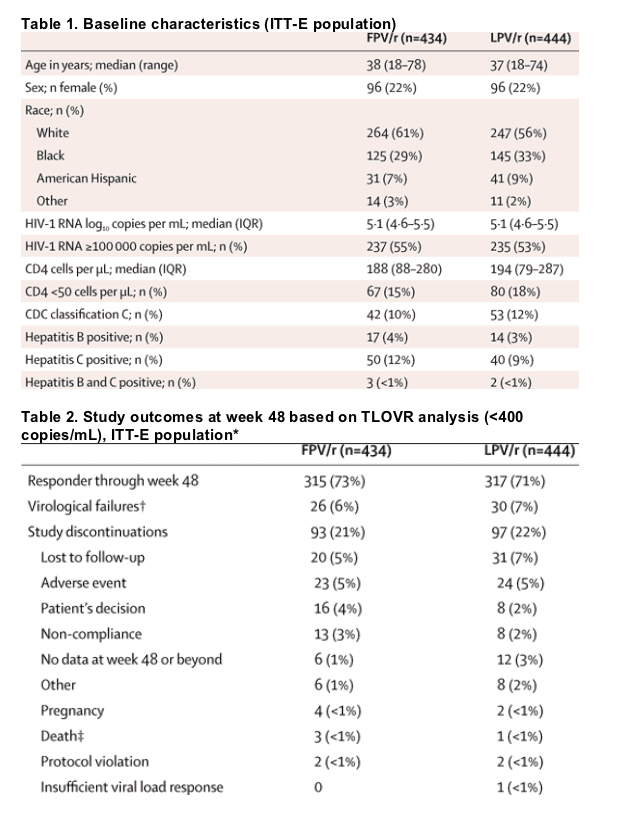
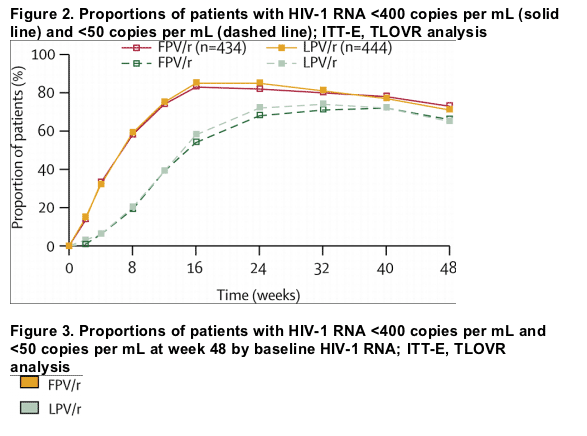
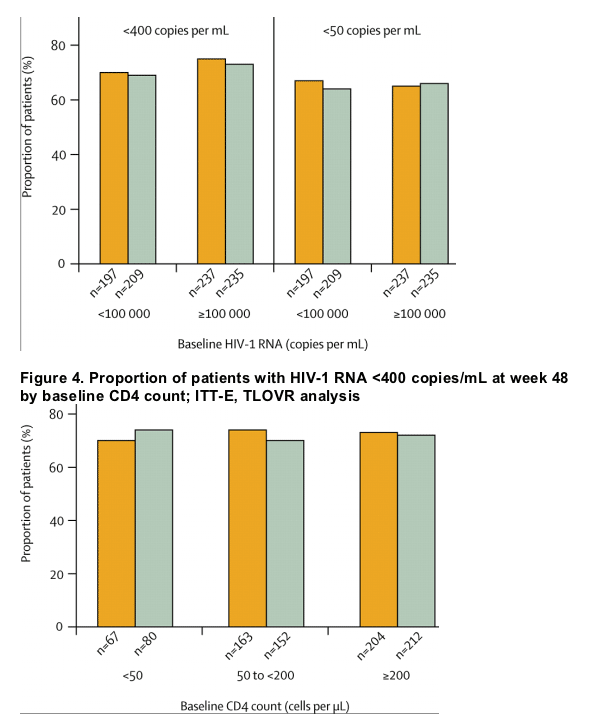
Study outcomes based on the TLOVR algorithm (HIV-1 RNA <400 copies per mL) are summarised in table 2. Treatment response assessed by baseline HIV-1 RNA strata or by baseline CD4 count indicated consistent results between groups and across strata (figures 3 and 4).
Immunological recovery was observed with a median CD4 count increase of 176 cells per μL in the fosamprenavir-ritonavir group (IQR 106-281) and 191 cells per μL in the lopinavir-ritonavir group (124-287) by week 48. Median CD4 counts at week 48 in the fosamprenavir-ritonavir group and lopinavir-ritonavir group were 375 cells per μL and 397 cells per μL, respectively. The proportion of patients who had disease progression to CDC class C or death was 11 of 434 (3%) for fosamprenavir-ritonavir and 11 of 444 (2%) for lopinavir-ritonavir. Five deaths were reported during the study period (four fosamprenavir-ritonavir, one lopinavir-ritonavir), none of which were related to study medication (table 2).
Drug-associated resistance as defined by International AIDS Society-USA criteria was rare in the 40 patients (5%) with protocol-defined virological failure (table 3). 35 of these patients had both baseline and on-treatment viral genotypes and phenotypes for analysis. Of these, 11 (31%) patients had virus with treatment-emergent mutations. Four (11%) acquired protease-inhibitor-associated mutations, all encoding mixtures of wild-type and mutant aminoacids including one I54I/L, two K20K/R, and one I62I/V, although none were major protease-inhibitor mutations. Evidence of genotypic resistance to NRTIs was also uncommon and occurred in seven (20%) of the 35 patients. No patient had virus that developed reduced phenotypic susceptibility to fosamprenavir-ritonavir or lopinavir-ritonavir (table 3). The median time to virological failure in the ITT-E population could not be estimated by the Kaplan-Meier method because of the small number of virological failures.
Median percentage adherence was similar for the protease-inhibitor components over the 48-week study period (fosamprenavir-ritonavir 98-4% vs lopinavir-ritonavir 98-0%). The median percentage adherence for abacavir-lamivudine was the same for both treatment arms (99-4%).
The safety population consisted of 879 patients (436 fosamprenavir-ritonavir, 443 lopinavir-ritonavir) and was based on treatment received. One patient randomised to fosamprenavir-ritonavir actually received lopinavir-ritonavir. Two patients randomised to lopinavir-ritonavir actually received fosamprenavir-ritonavir. Median exposure to all study medications was 48 weeks (range 0-1-62-7).
There were few premature discontinuations of study medication due to adverse events (53 [12%] fosamprenavir-ritonavir, 43 [10%] lopinavir-ritonavir). Adverse events of at least moderate severity occurred with similar frequency in the two treatment groups (table 4). Diarrhoea was the most common adverse event (exclusive of hypersensitivity reaction to abacavir) leading to treatment discontinuations and occurred in less than 3% of patients in both treatment groups (five [1%] fosamprenavir-ritonavir, seven [2%] lopinavir-ritonavir).
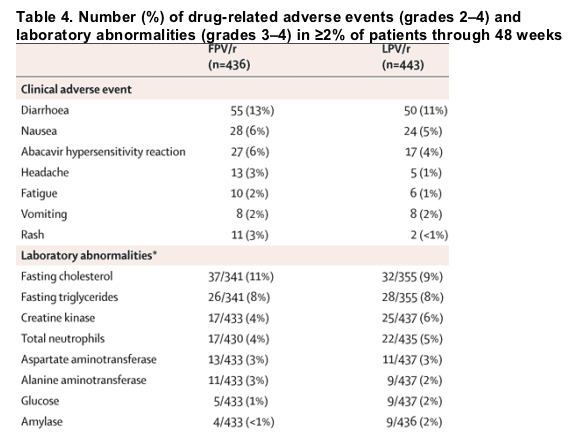
53 cases (6%) of suspected abacavir hypersensitivity were reported (32 [7%] fosamprenavir-ritonavir, 21 [5%] lopinavir-ritonavir), of which 2% were grade 3-4 for both groups. NRTI substitutions for abacavir included zidovudine, tenofovir, didanosine, and stavudine; study discontinuations for suspected abacavir hypersensitivity reactions were few (1%).
Treatment-emergent elevations in serum aminotransferases were more common in patients with evidence of co-infection with hepatitis B, hepatitis C, or both: grade 3-4 alanine transaminase elevations were observed in less than 1% (six of 738; four of 356 in fosamprenavir-ritonavir and two of 382 in lopinavir-ritonavir) of patients without co-infection compared with 12% (14 of 114; seven of 65 in fosamprenavir-ritonavir and seven of 49 in lopinavir-ritonavir) of patients co-infected with hepatitis B, C, or both, in both treatment groups.
Both groups showed similar changes from baseline in fasting lipid values at week 48 (table 5). Use of lipid-lowering medications was similar in the fosamprenavir-ritonavir and lopinavir-ritonavir groups (11%) during the study period.
Discussion
We undertook a large, well-powered comparison of two ritonavir-enhanced protease inhibitors, each combined with abacavir-lamivudine, in HIV-1 infected treatment-naive patients, and showed that fosamprenavir-ritonavir was non-inferior to lopinavir-ritonavir over 48 weeks. In fact, the antiviral and immunological responses in the fosamprenavir-ritonavir group were remarkably similar to those in the lopinavir-ritonavir group. Lopinavir-ritonavir is the only protease inhibitor currently preferred for initial treatment of HIV-1 by the US Department of Health and Human Services guidelines.3 At 48 weeks, using the FDA-mandated TLOVR analysis, 66% of all patients randomised had less than 50 copies per mL of HIV-1 RNA in the fosamprenavir-ritonavir group compared with 65% in the lopinavir-ritonavir group. This marked similarity was seen in all of the sensitivity analyses. Additionally, increases in CD4 count were robust and similar in the two treatment groups.
The potent antiretroviral effect of fosamprenavir-ritonavir was observed in all subsets of patients assessed, including those with high baseline HIV-1 RNA levels (≥100 000 copies per mL) and those with low baseline CD4 counts (<50 cells per μL), with little or no difference across subgroups or between treatment groups. Notably, before this study, lopinavir-ritonavir-based initial therapy had shown equally good virological outcomes in patients with low and high baseline HIV-1 RNA in a large randomised comparative trial.9 Fosamprenavir-ritonavir can now be added to the list of agents that can be administered with two nucleosides (including lamivudine or emtricitabine) for initial therapy and have similar potent responses in individuals with high viral loads and very low CD4 counts.
Tolerability and safety, numbers of treatment discontinuations, and increases in fasting cholesterol and triglycerides were similar between the groups. While rises in total cholesterol and triglycerides are clearly a consequence of most ritonavir-enhanced protease inhibitor therapies, the substantial increases in HDL cholesterol that are typically observed are consistent with the increase of about 40% in median HDL in this study.
Confirmed virological failure was uncommon in both treatment arms. Cross-study comparisons are limited, but the proportions of individuals with either virological rebound or detectable HIV-1 RNA with fosamprenavir-ritonavir are very similar to those reported in recent studies of lopinavir-ritonavir-based or efavirenz-based initial therapy.5,10-12 For example, 5% of individuals treated with tenofovir/emtricitabine and efavirenz had HIV-1 RNA levels of greater than 400 copies per mL with genotype results on or before 48 weeks in a large comparative study.10 Treatment-emergent drug resistance in patients with virological failure or rebound was also investigated in this study, and neither fosamprenavir-ritonavir nor lopinavir-ritonavir with two nucleosides selected for major protease-inhibitor-associated resistance mutations.13-14 These results confirm and extend those of previous studies that have shown a paucity of emergent protease-inhibitor resistance among first-line patients treated with combination therapy that included a ritonavir-boosted protease inhibitor.6,15,16 Additionally, emergence of nucleoside reverse transcriptase inhibitor-associated resistance mutations was similar between the groups and occurred in very few patients. Only three patients in the fosamprenavir-ritonavir group and four in the lopinavir-ritonavir group had virus with the M184V or I mutation associated with resistance to lamivudine or emtricitabine. These results contrast with those of studies of initial therapy with non-nucleoside reverse transcriptase inhibitors (NNRTI), in which virological failure was frequently associated with the acquisition of mutations associated with NNRTI resistance (about half of patients) and NRTI resistance (about 20-50% of patients).12-13,17
Although not a primary focus of the study, abacavir hypersensitivity reactions were well characterised, relatively uncommon, and consistent with previously described reports.18 Participants who had a suspected abacavir hypersensitivity reaction were allowed to substitute an alternative nucleoside analogue and remain on study; therefore, the proportion of individuals who discontinued the study due to hypersensitivity was small. Overall, abacavir-lamivudine was a well-tolerated backbone with both ritonavir-enhanced protease inhibitors. Excluding hypersensitivity reactions, there were few moderate to severe adverse events reported in either group.
Limitations of this study include its open-label design, which might bias assessment of tolerability or toxicity, but would be less likely to bias virological, immunological, or resistance endpoints. Additionally, the protease inhibitors in this study were administered twice daily, since both were not approved for once daily dosing when the study began.
Factors that affect selection of first-line antiretroviral therapy are many, but include the results of well done large randomised clinical trials. Our findings are likely to inform clinical decisions about choice of therapy for antiretroviral-naive HIV-1 infected individuals.
Methods
Participants
Patients were recruited from centres in the USA, Europe, and Canada and were eligible for enrollment if they were infected with HIV-1, aged 18 years or older, naive to antiretroviral therapy, and had HIV-1 RNA of 1000 copies per mL or greater. We also excluded patients if they had medical conditions or required medications that could compromise their safety or interfere with drug absorption, or if they had protocol-specified abnormal laboratory values. There were no exclusion criteria based on CD4-positive T-lymphocyte (CD4) counts.
The study was approved by the ethics review boards at each participating centre, and all patients provided written informed consent. This study was done in accordance with Good Clinical Practice.
Procedures
KLEAN was an open-label, randomised (1:1), non-inferiority study that compared fosamprenavir-ritonavir with lopinavir-ritonavir, each in combination with a fixed dose of abacavir-lamivudine, for 48 weeks. Site personnel called a centralised voice response system to randomise patients to either fosamprenavir 700 mg (one tablet) twice daily plus ritonavir 100 mg (one capsule) twice daily or the fixed dose of lopinavir-ritonavir 400 mg/100 mg (three capsules) twice daily, each in combination with abacavir-lamivudine 600 mg/300 mg (one fixed-dose combination tablet) once daily. Treatment allocation was based on a computer generated centralised randomisation schedule stratified by screening HIV-1 RNA (<100 000 copies per mL and ≥100 000 copies per mL) with a block size of four. Patients who had a suspected hypersensitivity reaction to abacavir were allowed to substitute any approved NRTI and continue in the study; no other antiretroviral substitutions were allowed.
Patients were evaluated at screening, at day 1 (baseline), and at weeks 2, 4, 8, 12, 16, 24, 32, 40, and 48, or withdrawal. At each visit, samples for HIV-1 RNA, CD4/CD8-positive lymphocyte subsets, clinical chemistries, and haematology were collected and analysed. Additionally, US Centers for Disease Control and Prevention (CDC) classification was assessed, and samples for hepatitis B and C serology and B-human chorionic gonadotropin (where appropriate) were collected and analysed at the baseline visit. HIV-1 RNA concentrations were measured by the Roche Cobas Amplicor HIV-1 Monitor test (version 1.5) at screening and baseline and by the Roche Cobas Amplicor HIV-1 Ultrasensitive Monitor test (version 1.5) post-baseline (Roche, Branchburg, USA). Samples exceeding the upper limit of the ultrasensitive assay were retested using the Amplicor HIV-1 Monitor (Roche). Adverse events, concurrent medications, HIV-associated conditions, and adherence based on pill counts were also assessed at each visit. Adverse events were graded with the 1992 Division of AIDS toxicity grading scale.8 A sample for viral genotypic and phenotypic analysis was stored at baseline and at all subsequent visits. Genotypic mutations were reported as any major or minor mutation from the International AIDS Society-USA Guidelines (November, 2005) emerging on treatment (compared with baseline) from patients meeting protocol-defined virological failure. Viral genotypes and phenotypes were assessed at Monogram BioSciences (South San Francisco, CA, USA). All other laboratory tests were done centrally by Quest Diagnostics (Van Nuys, CA, USA and Heston, UK).
The primary endpoints were the proportion of patients with HIV-1 RNA concentrations of less than 400 copies per mL at 48 weeks and the proportion who permanently discontinued randomised treatment due to adverse events. Secondary endpoints included the proportion of patients with HIV-1 RNA concentrations of less than 50 copies per mL, changes in HIV-1 RNA concentration and CD4 counts, time to virological failure, time to loss of virological response (TLOVR), development of genotypic and phenotypic resistance at virological failure, adherence based on pill counts, incidence of adverse events, and fasting lipid measures. Virological failure was defined as either a reduction of HIV-1 RNA to less than 400 copies per mL with a subsequent increase to 400 copies per mL or greater on two consecutive occasions, or a failure to achieve HIV-1 RNA less than 400 copies per mL by week 24. Patients experiencing virological failure could remain in the study if their HIV-1 RNA levels did not exceed 2000 copies per mL and at the investigator's discretion. To ensure thorough reporting, all suspected hypersensitivity reactions to abacavir were reported as serious adverse events, regardless of whether the event fulfilled the International Conference on Harmonization E2A definition of serious.
Statistical analysis
Assuming a 70% success rate in the fosamprenavir-ritonavir group and a 72% success rate in the lopinavir-ritonavir group, a total of 433 patients per arm provided 90% power to assess the non-inferiority of fosamprenavir-ritonavir twice daily compared with lopinavir-ritonavir twice daily at the one-sided 0-025 level of significance. Non-inferiority was defined as the lower bound of the two-sided 95% CI for the treatment difference being above -12%.
The intent-to-treat exposed (ITT-E) population included all patients exposed to at least one dose of randomized study medication. The primary efficacy analysis was based on the proportion of patients who achieved HIV-1 RNA of less than 400 copies per mL at 48 weeks (ITT-E, TLOVR). According to the TLOVR algorithm, an FDA-mandated analysis that categorises patients by treatment response, responders were patients with confirmed viral load less than 400 copies per mL on two consecutive occasions who had not yet met any non-responder criterion. Non-responders were patients who never achieved confirmed HIV-1 RNA less than 400 copies/mL on two consecutive occasions, prematurely discontinued study or protease inhibitor for any reason, had confirmed rebound to 400 copies per mL or greater, or had an unconfirmed HIV-1 RNA 400 copies per mL or greater on their final study visit. Additional analyses included ITT-E, observed, which included all observed data; ITT-E, missing/discontinuation=failure, in which missing data or data obtained after discontinuation of randomised study medication (fosamprenavir-ritonavir or lopinavir-ritonavir) were regarded as failures; and per-protocol, observed, which included all observed data for patients in the per-protocol population. This population included all randomised patients but excluded those who had major protocol deviations as identified by a blinded review. For the primary efficacy analysis and the corresponding sensitivity analyses, 95% CIs on the treatment difference were stratified by the baseline HIV-1 RNA concentration (<100 000 copies per mL or ≥100 000 copies per mL) using Mantel-Haenszel weights. The safety population included all randomised patients who consumed at least one dose of study drug and were analysed by the actual treatment received.
Percentage adherence was calculated by the total number of pills dispensed minus the total number of pills returned, divided by the total number of pills prescribed for the study duration. The median percentage adherence was calculated for fosamprenavir-ritonavir, lopinavir-ritonavir, and abacavir-lamivudine fixed-dose combination.
Role of the funding source
The sponsor developed the study design and analysis plan with input from prospective investigators. Decisions regarding the final protocol, data reviews, and publishing were made based on discussions between the sponsor and the study investigators. The corresponding author and the sponsor had full access to the data after official closure of the database. The corresponding author had final responsibility for the decision to submit for publication.
|
|
| |
| |
|
|
|