| |
Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial
|
| |
| |
The Lancet, Volume 366, Number 9493, 08 October 2005; 1279-1289
Prof John A Dormandy DSc a , et al. on behalf of the PROactive investigators
Summary
Background
Patients with type 2 diabetes are at high risk of fatal and non-fatal myocardial infarction and stroke. There is indirect evidence that agonists of peroxisome proliferator-activated receptor y (PPAR y) could reduce macrovascular complications. Our aim, therefore, was to ascertain whether pioglitazone reduces macrovascular morbidity and mortality in high-risk patients with type 2 diabetes.
Methods
We did a prospective, randomised controlled trial in 5238 patients with type 2 diabetes who had evidence of macrovascular disease. We recruited patients from primary-care practices and hospitals. We assigned patients to oral pioglitazone titrated from 15 mg to 45 mg (n=2605) or matching placebo (n=2633), to be taken in addition to their glucose-lowering drugs and other medications. Our primary endpoint was the composite of all-cause mortality, non fatal myocardial infarction (including silent myocardial infarction), stroke, acute coronary syndrome, endovascular or surgical intervention in the coronary or leg arteries, and amputation above the ankle. Analysis was by intention to treat. This study is registered as an International Standard Randomised Controlled Trial, number ISRCTN NCT00174993.
Findings
Two patients were lost to follow-up, but were included in analyses. The average time of observation was 34·5 months. 514 of 2605 patients in the pioglitazone group and 572 of 2633 patients in the placebo group had at least one event in the primary composite endpoint (HR 0·90, 95% CI 0·80-1·02, p=0·095). The main secondary endpoint was the composite of all-cause mortality, non-fatal myocardial infarction, and stroke. 301 patients in the pioglitazone group and 358 in the placebo group reached this endpoint (0·84, 0·72-0·98, p=0·027). Overall safety and tolerability was good with no change in the safety profile of pioglitazone identified. 6% (149 of 2065) and 4% (108 of 2633) of those in the pioglitazone and placebo groups, respectively, were admitted to hospital with heart failure (p=0.007); mortality rates from heart failure did not differ between groups.
Interpretation
Pioglitazone reduces the composite of all-cause mortality, non-fatal myocardial infarction, and stroke in patients with type 2 diabetes who have a high risk of macrovascular events.
Introduction
Patients with type 2 diabetes are at high risk of fatal and non-fatal macrovascular events. These events are the main reason for their decreased life expectancy, which is about 8 years shorter in a 40-year-old patient newly diagnosed with diabetes than in the general population.1 There is a two-fold to four-fold increased risk of a macrovascular event in patients with, compared with those without, diabetes.2,3 Haffner and colleagues4 noted that the risk of a cardiovascular complication in a patient with diabetes was similar to that of a patient without diabetes who had had a myocardial infarction. In the Heart Protection Study,5 patients with diabetes and a history of cardiovascular disease at entry had almost a three-fold higher risk of a new cardiovascular event than did those without such a history.
Intensive control of glycaemia decreases microvascular complications, such as retinopathy and nephropathy, but has no great effect on macrovascular complications or all-cause mortality. However, in the UK prospective diabetes study (UKPDS),6 findings of a retrospective analysis in a subgroup of 342 overweight patients who received metformin showed a significant decrease in cardiovascular disease and total mortality.
Pioglitazone is an agonist of peroxisome proliferator-activated receptor _ (PPAR _) used to treat type 2 diabetes.7 The overall pattern of changes induced by pioglitazone suggests a general improvement in various risk factors that might reduce cardiovascular morbidity and mortality. Additionally, pioglitazone reduces the levels of various inflammatory markers, such as highly sensitive C-reactive protein (hsCRP), independently of its effect on glycaemic control.8
Our aim was to ascertain whether pioglitazone reduces cardiovascular morbidity and mortality in patients with type 2 diabetes, and to assess the safety and tolerability of such treatment.
Methods
Patients
The PROactive (PROspective pioglitAzone Clinical Trial In macroVascular Events) protocol has been described in detail previously.9 Between May, 2001, and April, 2002, we recruited patients from primary-care practices and diabetic or cardiovascular specialist departments in hospitals to a randomised controlled trial. We included patients with type 2 diabetes who were aged 35-75 years if they had an haemoglobin A1c (HBA1c) concentration greater than the local laboratory equivalent of 6·5% for a Diabetes Control and Complications Trial-traceable assay (DCCT), despite existing treatment with diet alone or with oral glucose-lowering agents with or without insulin. Patients also had to have evidence of extensive macrovascular disease before recruitment, defined by one or more of the following criteria: myocardial infarction or stroke at least 6 months before entry to the trial, percutaneous coronary intervention or coronary artery bypass surgery at least 6 months before recruitment, acute coronary syndrome at least 3 months before recruitment, or objective evidence of coronary artery disease or obstructive arterial disease in the leg. Objective evidence of coronary artery disease was defined as a positive exercise test, angiography showing at least one stenosis of more than 50%, or positive scintigraphy. Obstructive arterial disease of the leg was defined as a previous major amputation or intermittent claudication with an ankle or toe brachial pressure index of less than 0·9.
We excluded patients if they: had type 1 diabetes; were taking only insulin; had planned coronary or peripheral revascularisation; had New York Heart Association class II heart failure or above; had ischaemic ulcers, gangrene, or rest pain in the leg; had had haemodialysis; or had greater than 2·5 times the upper limit of normal concentrations of alanine aminotransferase.
All patients provided written informed consent. The study protocol was approved by local and national ethics committees and regulatory agencies, and was done in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines.
Procedures
We randomly assigned patients to oral pioglitazone or matching placebo in addition to their existing medication(s) for diabetes. Study medication was assigned via a central interactive voice response system. Allocation of patients to treatment groups was done by the method of randomised permuted blocks within centre. All investigators and study personnel were unaware of treatment assignment for the duration of the study. Only the data and safety monitoring committee saw unblinded data, none of whom had any contact with the study participants. The randomisation sequence was generated by a member of the Statistics Department of Nottingham Clinical Research Limited. Once these lists had been checked, all files were passed on to the interactive voice response system coordinator, who maintained these files securely for the duration of the trial. The original lists were deleted by the Department of Statistics, who had no access to the randomisation code until the study was unblinded. Masking of drugs was achieved by using matching placebo.
If allocated, we gave patients oral pioglitazone 15 mg for the first month, 30 mg for the second month, and 45 mg thereafter to achieve the maximum tolerated dose, according to the licensed dose range for pioglitazone. At any time during the study, the dose of study drug could be adjusted within the same dose range if clinically indicated. Throughout the study, investigators were required to increase all therapy to an optimum, according to the International Diabetes Federation European Region 1999 guidelines.10 We drew particular attention to the need to reach an HBA1c concentration below the recommended target (<6·5%) and to increase to an optimum lipid-altering, antiplatelet, and antihypertensive therapy.
We saw patients monthly for the first 2 months, then every 2 months for the first year, and thereafter every 3 months until the final visit. We followed-up all patients until the end of the study even if they permanently ceased study medication before the study end. We measured vital signs and bodyweight at every visit. We obtained standard 12-lead electrocardiograms at the beginning of the study, at yearly intervals thereafter, and at the final visit. Two independent reviewers assessed all electrocardiograms for evidence of silent myocardial infarction on behalf of the endpoint adjudication committee. We took blood samples at baseline for central laboratory assessment of concentrations of HBA1c, triglyceride, HDL cholesterol, LDL cholesterol, alanine aminotransferase, aspartate aminotransferase, total bilirubin, alkaline phosphatase, and creatinine. Thereafter, we measured HBA1c, fasting lipid, and creatinine concentrations every 6 months, and liver function at every visit in the first year and every 6 months in subsequent years. Urinary albumin concentration was measured locally at the beginning and at the end of the study, using Micral Test strips (Roche Diagnostics, Mannheim, Germany). We identified the presence of retinopathy, nephropathy, and neuropathy from the patients' records. Blood pressure was measured with routine clinical methods.
All samples were measured in a central laboratory that participated in the appropriate national quality-control schemes for all analyses. We measured HDL-cholesterol and LDL-cholesterol concentrations with direct quantitative enzymatic methods, and triglyceride levels with a glycerol-blanked, enzymatic assay. Methods used to measure concentrations of HDL-cholesterol and triglycerides were accredited by the Centres for Disease Control Lipid Standardisation Program. We undertook all central laboratory methods on automated Hitachi (Tokyo, Japan) P-Modular platforms, using Roche reagents (Roche Diagnostics, Mannheim, Germany), with standards and controls as recommended by the manufacturer. We measured HBA1c concentrations in whole blood with a BIO-RAD-Variant ion exchange high-pressure liquid chromatography analyser (Biorad, Hercules, CA, USA), with standards and controls supplied by the manufacturer. The upper limit of normal for the laboratory was 6·4%.
Our primary endpoint was time from randomisation to: all-cause mortality, non-fatal myocardial infarction (including silent myocardial infarction), stroke, acute coronary syndrome, endovascular or surgical intervention on the coronary or leg arteries, or amputation above the ankle. We diagnosed a non-fatal myocardial infarction if the patient survived more than 24 h from onset of symptoms and, in the absence of percutaneous coronary intervention or coronary artery bypass graft, had at least two of: symptoms suggestive of myocardial infarction (ischaemic chest pain or discomfort) lasting 30 min or longer, electrocardiographic evidence of myocardial infarction, or raised cardiac serum markers; or after percutaneous coronary intervention or coronary artery bypass graft the patient had electrocardiographic evidence of myocardial infarction. Silent myocardial infarction was defined as new Q waves on two contiguous leads or R-wave reduction in the precordial leads without a change in axis deviation. Acute coronary syndrome was noted if the patients received treatment in hospital for ischaemic discomfort at rest that lasted at least 5 min and had electrocardiographic changes or raised cardiac serum markers not sufficiently high to indicate myocardial infarction, or both. Coronary revascularisation was when a patient underwent percutaneous transluminal coronary intervention-eg, angioplasty, stenting, atherectomy, laser ablation-or coronary artery bypass graft. Stroke was defined as acute focal neurological deficit lasting for longer than 24 h or resulting in death within 24 h of the onset of symptoms, which was diagnosed as being due to cerebral lesion of vascular origin but excluding subarachnoid haemorrhage. Major leg amputation included all amputations of the leg above the ankle. Revascularisation in the leg was noted if a patient underwent any of surgical bypass, atherectomy, angioplasty, or thrombolysis.
The prespecified secondary endpoints, in order of priority, were: time to the first event of death from any cause, myocardial infarction (excluding silent myocardial infarction), and stroke (main secondary endpoint in rest of this report); cardiovascular death; and time to individual components of the primary composite endpoint. We classified all fatal events as cardiovascular unless there was a clear non-cardiovascular cause.
We reported all potential endpoints and other serious adverse events to the coordinating centre within 1 working day of becoming aware of the event. We defined serious adverse events as: resulting in death, life-threatening, needing or prolonging in-patient admission, resulting in persistent or significant disability, or needing intervention to prevent any of the above. We elicited non-serious adverse events at every visit. Investigators were required to report, in particular, occurrences of symptoms compatible with hypoglycaemia, heart failure (as judged by the investigator), and oedema in the absence of heart failure, plus any adverse event leading to discontinuation of the study drug.
Monitors reviewed patients' records regularly to ensure that all potential endpoints and other serious adverse events were being reported. All reports of serious adverse event were checked against the patients' clinical notes. An independent panel, working with the endpoint adjudication committee, assessed all potential endpoints and classified them in accord with predefined criteria. The study data and safety monitoring committee supervised the study and assessed unblinded data to ensure the continued safety of participants throughout.
Nottingham Clinical Research Group acted as a coordinating centre, providing project management, data management, central randomisation services, and statistical analysis. ICON Clinical Research managed and monitored the sites, and did central laboratory measurements.
Statistical analysis
Our planned study sample size of 5000 patients was based on the assumptions of a 6% annual primary event rate in the placebo group, recruitment of patients over 18 months, and a total trial duration of 4 years. A time-to-event analysis was planned, and thus the study had 91% power to detect a 20% reduction in the hazard with a type I error of 0·05. To maintain this power, all patients had to be followed-up until at least 760 patients had one endpoint event or more.
Since the event rate was higher than expected and the enrolment rate was faster than planned, the mean duration of exposure would have been shorter than originally anticipated. Therefore, to ensure sufficient duration of exposure, the protocol was amended in May, 2003, to specify that the trial should continue until the last patient recruited had been followed-up for 30 months and at least 760 patients had had one or more endpoint events.
Two pre-planned interim analyses were done by the data and safety monitoring committee when about half and three-quarters of the target number of endpoints had been reached. We controlled the type I error with the method of Lan and Demets with the O'Brien-Fleming alpha spending function.11 The final analysis of the primary endpoint thus needed the observed significance level (two-sided) to be less than 0·044 for the treatment difference to be declared significant at the 5% level.
All time-to-event analyses were done by fitting a proportional hazards survival model with treatment as the only covariate. The proportional hazards assumption was tested with the method described by Grambsch and Therneau.12 Homogeneity of response was examined by testing for interaction in each of 25 prespecified sets of subgroups. We used linear models or logistic regression models for other endpoints, as appropriate. All analyses were by intention to treat.
This study is registered as an International Standard Randomised Controlled Trial, number ISRCTN NCT00174993.
Role of the funding source
The study was designed by the international steering committee, who also approved the protocol and amendments. The sponsors had two representatives on the international steering committee and the same two were also members of the executive committee. Data analysis, data interpretation, and writing of the report was done by the executive committee, with contributions from the international steering committee, the data and safety monitoring committee, and the endpoint adjudication committee. All the authors had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Results
Figure 1 shows the trial profile. 5238 patients from 321 centres in 19 European countries were randomly assigned to either pioglitazone (n=2605) or placebo (n=2633); 1681 patients were recruited from the community and 3557 from hospitals. All patients commenced study medication and all received their intended treatment. 16% of patients assigned pioglitazone and 17% of those assigned placebo discontinued study medication before death or final visit (figure 1). We completed final visits between November, 2004, and January, 2005. The average time of observation was 34·5 months. Two patients were lost to follow-up. All other patients were followed-up to their final visit or death. The treatment code was broken for three patients (all placebo) during the study for medical or medicolegal reasons.
The two groups were well matched with respect to baseline characteristics (table 1). Mean age overall was 61·8 years, with the median time since diagnosis of diabetes being 8 years. At randomisation, 62% of patients were taking metformin and 62% were taking a sulphonylurea either as monotherapy or in combination for diabetes control. More than 30% of patients were on insulin. Contrary to the study entry criteria, 13 patients (0·2%) had insulin as their only glucose-lowering medication.
Table 2 shows details of macrovascular disease and related concomitant medications taken. Patients had a high level of previous morbidity. We randomised 82 patients (2%) who we subsequently noted did not meet any of the strictly defined criteria for entry based on macrovascular history. Of these, 20 patients did not have any documented evidence of a previous macrovascular event. We included all 82 patients in all intention-to-treat analyses, but assigned them to the so-called absent subgroup for each of the subgroup analyses that related to macrovascular entry criteria.
Throughout, pioglitazone was well tolerated, with 89% (2235 of 2521) of patients reaching the 45 mg dose at the 2-month visit compared with 91% (2293 of 2517) of matching placebo. Thereafter, at least 93% of patients continuing on pioglitazone received the highest dose compared with at least 95% of those on placebo. Compliance in both treatment groups, as defined by more than 75% of tablets used, was greater than 95%.
Figure 2 shows Kaplan-Meier estimates of the proportion of patients reaching an event within the primary composite endpoint by treatment. Fewer patients in the pioglitazone group had at least one event than in the placebo group, though this finding was not significant. Figure 3 shows the Kaplan-Meier estimate of the proportion of patients reaching the main secondary endpoint of all-cause mortality, non-fatal myocardial infarction (excluding silent myocardial infarction), or stroke. Fewer patients in the pioglitazone than in the placebo group had at least one event. The difference was significant. There was no significant violation of the proportional hazards assumption (p=0·085 for the primary endpoint and p=0·616 for the main secondary endpoint). Table 3 shows the breakdown of event types within the primary and the main secondary endpoints. The four most frequent component endpoints were death, myocardial infarction, stroke, and coronary revascularisation. All are well represented in the primary composite endpoint, and the first three constitute the main secondary endpoint. There were 127 cardiovascular deaths in the group treated with pioglitazone compared with 136 in the placebo group. There were 50 non-cardiovascular deaths in each group.
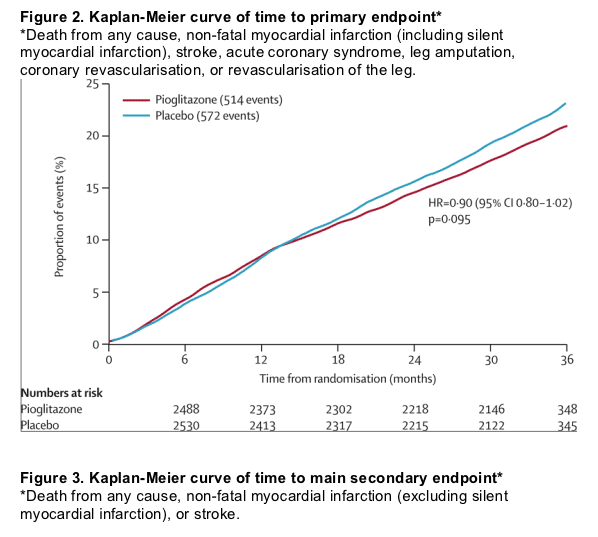
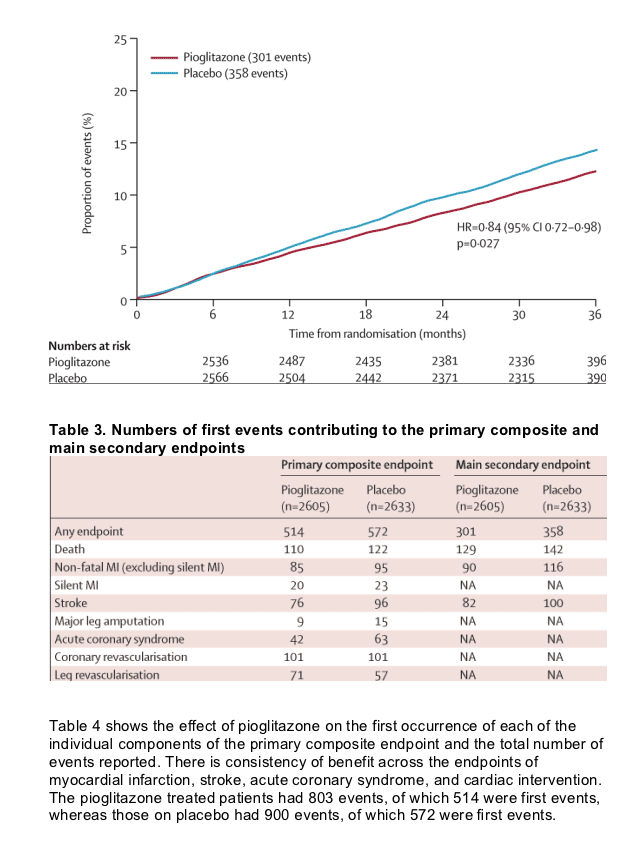
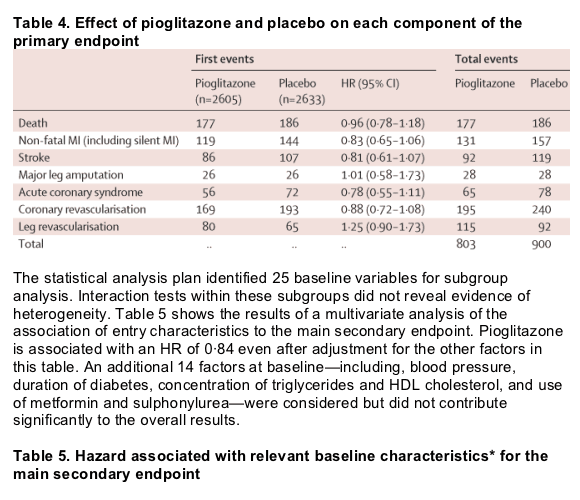
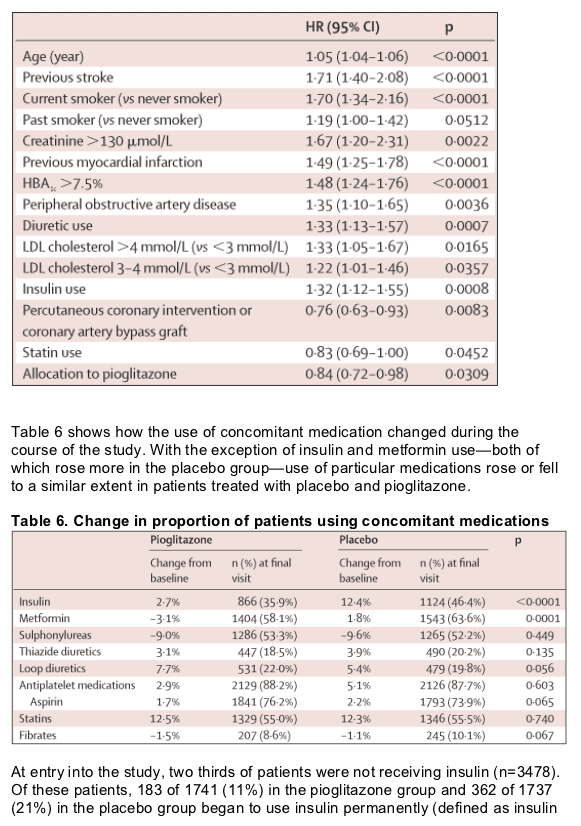
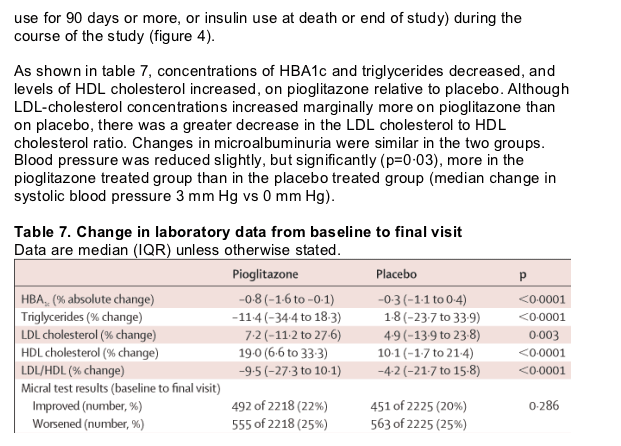
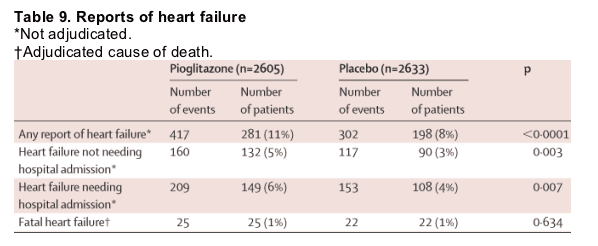
Table 8 summarises the incidence of serious adverse events that arose in more than 1% of patients. There were fewer serious adverse events in the pioglitazone group than in the placebo group, this difference indicating both the lower incidence of endpoint events and fewer other serious events. Table 9 shows the reporting rates of heart failure in the study. Despite the increase in reported heart failure in the pioglitazone group, the number of deaths from heart failure was similar in each group. Furthermore, 903 patients reported oedema without heart failure (562 pioglitazone, 341 placebo). Symptoms compatible with hypoglycaemia arose in 726 (28%) patients on pioglitazone and 528 (20%) on placebo, (p<0·0001) whereas hypoglycaemia that resulted in admission to hospital arose in 19 and 11 patients, respectively (p=0·14). Slightly more patients in the placebo group needed to be admitted for management of their diabetes. Overall, fewer patients who received pioglitazone were admitted to hospital than those on placebo (1145 [44%] vs 1217 [46%]). There was no difference in the overall incidence of malignant neoplasms. There were some imbalances in the incidence of individual tumours. There were more bladder tumours (14 vs six) and fewer cases of breast cancer (three vs 11) reported in the pioglitazone group compared with placebo. We noted no cases of acute liver toxicity, although there was a small reduction (median 5%, IQR -27 to 20) in the alanine aminotransferase levels in the pioglitazone group compared with a small increase (8%, -17 to 38) in the placebo group. Increases of alanine aminotransferase to more than three times the upper limit of normal at any time during the study arose in 20 pioglitazone-treated and 33 placebo-treated patients. Creatinine values remained constant in both groups throughout the study. There was a 3·6 kg increase in mean bodyweight (range -30 to 29) in the pioglitazone group and a 0·4 kg decrease (-36 to 33) in the placebo group (p<0·0001).
Discussion
Our findings show that pioglitazone non-significantly reduces the risk of the composite primary endpoint-death from any cause, non-fatal myocardial infarction (including silent myocardial infarction), stroke, acute coronary syndrome, leg amputation, coronary revascularisation, or revascularisation of the leg. The pre-defined main secondary endpoint-all-cause mortality, myocardial infarction, or stroke-was also reduced, significantly, in the pioglitazone group. Kaplan-Meier estimates indicate that allocation of 1000 patients to pioglitazone would avoid 21 first myocardial infarctions, strokes, or deaths over 3 years. In other words, 48 patients would need to be treated for 3 years to avoid one first major cardiovascular event. This finding, however, might be an underestimate of the benefit of pioglitazone, since events subsequent to the initial one are also reduced. It is noteworthy that this improvement in outcome arose on top of normal medical care, which included glucose-lowering, antiplatelet, antihypertensive, and lipid-altering therapies. Furthermore, the improvement was seen in a group of particularly ill patients who we selected on the basis of a macrovascular history.
When the protocol was devised, we thought that the need for amputation, or cardiac or leg revascularisation, was likely to indicate macrovascular deterioration and would respond to therapy in a similar way to stroke and myocardial infarction. This hypothesis did not prove correct in the case of cardiac and leg revascularisation, perhaps because these endpoints are in part determined by the decision to intervene being based on local surgical or medical practice. All three outcomes of the main secondary endpoint were improved. The number of patients reporting an event that are discounted by moving from the primary to the principal secondary endpoint is the same (213, 214) in each group.
Glycaemic control was better in the pioglitazone group than in the placebo group, despite an increased use of metformin and insulin in the placebo group; dyslipidaemia improved without any difference in the use of lipid-altering agents. There was a small increase in LDL-cholesterol concentrations in the pioglitazone group, but the ratio of LDL cholesterol to HDL cholesterol improved more than on placebo. The increase in LDL-cholesterol concentrations could be related to a change in the distribution of LDL particles. Total LDL particles are reduced with pioglitazone.13 Therefore, the increase in concentrations of LDL cholesterol might not be considered adverse.
How pioglitazone improved cardiovascular outcome in our patients is unclear. The pioglitazone-treated group had a better metabolic profile in terms of glucose, HDL cholesterol, and triglyceride concentrations, and a better blood-pressure profile at the end of the study than at the beginning. The improvement in glycaemic control arose despite the fact that investigators were urged to adhere to the 1999 International Diabetes Federation guidelines and targets for the management of their patients and could alter background medication. Indeed, this requirement explains in part the increased use of insulin and metformin in the placebo group. The improvement in concentrations of triglycerides and HDL cholesterol are also of significant magnitude, and might have contributed to the outcome. The difference in LDL-cholesterol concentrations between the groups is unlikely to be of clinical significance. Although small, the difference in blood pressure between the groups might, however, have contributed to the outcome. Reaven14 has proposed that insulin resistance is the link between hyperglycaemia, dyslipidaemia, hypertension, and macrovascular disease. Thiazolidinediones, such as pioglitazone, improve insulin sensitivity through their effect on the PPAR _ receptor. This mechanism could be the link between treatment and reduced risk of macrovascular disease in patients with diabetes, but further work is needed to confirm this notion.
We also noted a reduced need to start taking insulin while on pioglitazone compared with placebo. The hazard reduction of 50% could indicate that doctors treating patients in the control group, who were unable to prescribe pioglitazone, used insulin instead to try to improve glycaemic control. Alternatively, pioglitazone might reduce the concentration of glucose in the blood to below a threshold at which insulin would be used. Finally, as previously suggested, pioglitazone could have a specific _-cell sparing effect, manifest in other clinical studies by a reduction of circulating insulin,15 and in animal studies by regranulation of the _ cell.16
We believe our results are generalisable to all patients with type 2 diabetes. We recruited patients from 19 countries in Europe; both from primary-care and secondary-care settings. Individuals were at high risk of macrovascular events by virtue of the entry criteria, which required evidence of macrovascular disease. Furthermore, patients were on a wide range of glucose-lowering medications, including insulin. The beneficial effects of pioglitazone are apparent in patients who take insulin as well as in those who do not, and are independent of the use of other oral glucose-lowering treatments. Our results should also be applicable to patients who have not had a macrovascular event, since virtually all patients with type 2 diabetes develop atherosclerotic disease and there is a two-fold to four-fold increased risk in those with, compared to those without, diabetes. Since our subgroup analyses did not reveal any great heterogeneity across the 25 variable categories (a total of 56 subgroups), the overall estimate of efficacy provides the best estimate of effect for all subgroups.
The results of the Universities Group Diabetes Programme17 and UKPDS18 indicated no clear improvements in cardiovascular outcomes after an intensive blood glucose-lowering regimen in patients newly diagnosed with type 2 diabetes. Findings of a subsequent analysis6 of patients in UKPDS who were obese and who took metformin as the main treatment for their diabetes rather than conventional, non-intensive therapy, showed a significant improvement in macrovascular outcomes. However, in obese patients given metformin as an adjunct to sulphonylurea there was a non-significant, increase in cardiovascular events.
Compared with placebo, we noted no excess deaths in the pioglitazone group, and identified no liver toxicity. Slightly fewer patients in the pioglitazone group reported non-endpoint serious adverse events than in the placebo group. Consistent with the reported side-effect profile for pioglitazone, there was an increased rate of oedema and heart failure, though mortality due to heart failure did not differ between groups. The increased reporting of heart failure in the pioglitazone group might, at least in part, indicate a diagnostic bias because of the increased oedema in the pioglitazone group. It is noteworthy that heart failure was not a centrally adjudicated event. The adverse-event profile was otherwise unremarkable.
The data and safety monitoring committee reviewed the 20 bladder cases with external experts (S Cohen, University of Nebraska Medical Center, and D Phillips, UK Institute of Cancer Research) before the study was unblinded. The experts considered that the 11 tumours that occurred within 1 year of randomisation (eight pioglitazone, three placebo) could not plausibly be related to treatment. After unblinding, there remained nine cases: six and three cases in the pioglitazone and placebo groups, respectively. Of these, four and two cases had known risk factors in their history (smoking, exposure to potential carcinogens, family history, previous tumour, urinary tract infection). Taking into account the timeframe of these cases and the potential confounding factors, it is improbable that the imbalance is related to pioglitazone treatment.
In summary, in patients with type 2 diabetes who are at high cardiovascular risk, pioglitazone improves cardiovascular outcome, and reduces the need to add insulin to glucose-lowering regimens compared with placebo.
Contributors
All authors helped to devise the study protocol, reviewed the full data, and commented on the draft manuscript. J A Dormandy (Study Chairman) chaired the Protocol and Executive Committee and participated in writing all drafts of the manuscript. D J A Eckland initiated the study, chaired the Operations Committee, and contributed to all drafts of the final manuscript. I K Moules chaired the Operations Committee and contributed to all drafts of the final manuscript. A M Skene was responsible for project and data management and statistical analyses. M H Tan contributed to all drafts of the final manuscript. B Charbonnel, E Erdmann, and M Massi-Benedetti were members of the Executive Committee supervising the day-to-day running of the study and writing the initial, working, and final draft of the manuscript. P J Lefebvre chaired the Data and Safety Monitoring Committee and was primarily responsible for drafting the safety section of the manuscript. G D Murray gave independent statistical advice and contributed to the final manuscript. E Standl, R Wilcox, and L Wilhelmsen served on the Data and Safety Monitoring Committee and contributed to the safety and other sections of the final manuscript. J Betteridge, K Birkeland, B Charbonnel, A Golay, R J Heine, L Koranyi, M Laakso, M Massi-Benedetti, M Moka_, A Norkus, V Pirags, T Podar, A Scheen, W Scherbaum, G Schernthaner, O Schmitz, J _krha, U Smith, and J Tato_ were members of the International Steering Committee, helped devise the study protocol, and supervised the study in their respective countries, and contributed to drafts of the final manuscript.
Conflict of interest statement
J A Dormandy, B Charbonnel, E Erdmann, M Massi-Benedetti, E Standl, R G Wilcox, L Wilhelmsen, J Betteridge, K Birkeland, A Golay, R J Heine, L Koranyi, M Laakso, M Moka_, A Norkus, V Pirags, T Podar, A Scheen, W Scherbaum, G Schernthaner, O Schmitz, J _krha, U Smith, and J Tato_ have served as consultants to, and received travel expenses and payments for speaking at meetings from, Takeda. D J A Eckland was an employee of, and has served as a consultant to Takeda. I K Moules works for Takeda. M H Tan works for Eli Lilly and Company. A M Skene is the Managing Director of Nottingham Clinical Research Group, which was contracted by Takeda. The University of Liege (International Diabetes Federation account) was compensated for the work done by P J Lefebvre as chairman of the data and safety monitoring committee. The independent statistical group located at the University of Edinburgh Medical School was compensated for the work done by G D Murray, statistician and Director of the independent statistical group.
|
|
| |
| |
|
|
|