| |
Partially Suppressive HAART Risks Future Drug Options
|
| |
| |
Rate of Viral Evolution and Risk of Losing Future Drug Options in Heavily Pretreated, HIV-Infected Patients Who Continue to Receive a Stable, Partially Suppressive Treatment Regimen
Clinical Infectious Diseases Nov 15, 2006;43:1329-1336
Hiroyu Hatano,1 Peter Hunt,1 Jodi Weidler,2 Eoin Coakley,2 Rebecca Hoh,1 Teri Liegler,1,3 Jeffrey N. Martin,1 and Steven G. Deeks1
1University of California San Francisco, 2Monogram Biosciences, and 3Gladstone Institute of Virology and Immunology, San Francisco, California
The goal of combination antiretroviral therapy is to prevent HIV disease progression over the course of a normal lifespan. Theoretically, this long-term goal will only be achieved if a treatment regimen can be constructed that decreases virus replication to a level that does not permit ongoing viral evolution or the emergence of drug resistance mutations. Thus, for patients experiencing incomplete viral suppression while receiving a combination regimen, current clinical guidelines recommend switching therapy to a new, potentially fully suppressive treatment regimen whenever possible [1, 2]. However, because of issues regarding cross-resistance, drug toxicity, and cost, many treatment-experienced patients do not have access to a fully effective "salvage" regimen. Because interrupting all therapy for such patients is associated with an increased risk of clinical progression [3], these patients often continue to receive a stable regimen with the goal of preventing disease progression until at least 2-and, preferably, 3-fully effective drug options become available.
The major risk of maintaining a partially suppressive treatment regimen is the continued emergence of mutations that reduce future drug options. Several studies have attempted to estimate this risk using genotypic resistance data generated in the course of routine clinical care [4, 5]. However, no study has addressed this issue in a prospective manner using both genotypic and phenotypic resistance tests performed at predetermined intervals. Therefore, we conducted the following study to evaluate the risk of waiting to switch therapy in treatment-experienced, HIV-infected individuals who continue to receive a partially suppressive treatment regimen.
"....In this study, we measured the risk of developing drug-resistance mutations over time in a cohort of treatment-experienced, HIV-infected patients who continued to receive (at the discretion of their primary care providers) a stable antiretroviral regimen despite incomplete viral suppression. An estimated 44% of patients developed at least 1 new drug resistance mutation over 1 year, whereas 30% lost the phenotypic equivalent of 1 susceptible drug at 1 year..... The resistance cost of maintaining a stable regimen despite incomplete viral suppression argues for aggressive modification of therapy during virologic failure. However, for many patients, a fully suppressive option is not available, and any decision to switch therapy early risks the rapid loss of future drug options..... The risk of losing future drug options is likely to be the major risk associated with maintaining a partially suppressive regimen. Although we observed a significant risk of viral evolution overall, the risk of accumulating genotypic resistance to tipranavir and darunavir was relatively low.....In the setting of relative clinical stability, it may be prudent to wait to switch therapy until at least 2 new effective antiretroviral agents are available."
ABSTRACT
Background. Many treatment-experienced, HIV-infected patients who have limited therapeutic options for complete viral suppression continue to receive a partially suppressive treatment regimen pending the availability of at least 2 new antiretroviral drugs. The major risk of this approach is ongoing viral evolution and the loss of future drug options.
Methods. Antiretroviral-treated subjects with incomplete viral suppression were sampled from a clinic-based cohort. Inclusion criteria were receipt of a stable treatment regimen for 120 days, a plasma HIV RNA load of >500 copies/mL, and >1 resistance mutation. Phenotypic and genotypic resistance testing was performed every 4 months.
Results.
The 106 patients who were eligible for the study had a median of 3 observations during a median of 11.3 months.
An estimated 23% and 18% developed at least 1 new nucleoside analogue and 1 new protease inhibitor mutation at 1 year, respectively.
An estimated 30% lost the phenotypic equivalent of 1 susceptible drug at 1 year. A lower number of total mutations at baseline was a significant predictor of developing a new nucleoside analogue mutation (P = .01). At 1 year, the probability that an existing mutation would become undetectable using population-based sequencing was 32%. There was a higher rate of change at nonresistance codons than at codons known to be associated with drug resistance.
Conclusions. Heavily pretreated patients with HIV infection who remain on a partially suppressive regimen have a measurable risk of losing future drug options, particularly those patients who have few baseline mutations. Resistance mutations vary over time, which suggests that the results of any single resistance test may not be representative of all mutations selected by a given treatment regimen.
METHODS
Study design. This was an observational cohort study of patients enrolled in the SCOPE cohort. SCOPE is an ongoing, clinic-based prospective study of >600 subjects that is focused on defining the immunologic and virologic characteristics associated with disease outcome in chronically HIV-infected individuals. Most subjects are recruited from the HIV clinic at San Francisco General Hospital or the San Francisco Veterans Affairs Medical Center. At enrollment, 50% of the cohort had measurable plasma HIV RNA levels while receiving combination antiretroviral therapy. All SCOPE participants are evaluated every 4 months, at which time a detailed health questionnaire is completed and biological specimens are collected and stored. All treatment decisions are made by primary care providers.
Study participants. Individuals from the SCOPE cohort were eligible for this study if they met the following characteristics at any time: (1) a stable antiretroviral therapy regimen for at least 120 days, (2) at least 2 plasma HIV RNA levels >100 copies/mL in the preceding 90 days, (3) a plasma HIV RNA level >500 copies/mL at the baseline visit for this study, (4) evidence of at least 1 major or minor genotypic resistance mutation (excluding L63P) at baseline, and (5) at least 1 follow-up visit. Observations were censored at the time of any treatment modification (change or discontinuation of antiretroviral drugs) or if the plasma HIV RNA level decreased to <75 copies/mL. For subjects who met inclusion criteria on multiple occasions, their most recent episode was evaluated. All subjects provided written, informed consent. This study was approved by the University of California San Francisco Committee on Human Research.
Study measurements. Genotypic and phenotypic resistance testing was performed every 4 months. Resistance testing was performed at Monogram Biosciences (South San Francisco, CA). Genotypic resistance measurements were performed using the GeneSeq assay (Monogram Biosciences), as described in detail elsewhere [6].
Phenotypic susceptibility to antiretroviral drugs was determined using a recombinant, virus-based assay that measures the fold-change in drug concentration necessary to inhibit 50% of the drug-resistant variant (IC50), compared with a wild-type reference strain (PhenoSense HIV; Monogram Biosciences). For analytic purposes, phenotypic susceptibility was based on lower and upper clinical "cutoffs" that were estimated for the interpretation of this assay (Michael Bates, Monogram Biosciences, personal communication) [7]. A phenotypic susceptibility score (PSS) was calculated for the antiretroviral regimen that the subject was receiving during the study. A score of 1 was assigned if the drug was fully active (fold-change in IC50 below the lower cutoff). A score of 0.5 was assigned if the drug was partially effective (fold-change in IC50 between the lower and upper cutoffs). A score of 0 was assigned if the drug had no clear activity (fold-change in IC50 above the upper cutoff). The PhenoSense assay (Monogram Biosciences) was also used to measure replication capacity. Plasma HIV RNA levels were determined using the branched DNA amplification technique (Quantiplex HIV RNA, version 3.0 [Chiron Corporation]; lower limit of detection, 75 copies/mL).
Genotypic resistance end points. Primary study end points were time to development of 1 new nucleoside analogue mutation (using the most recent International AIDS Society-USA [IAS-USA] guidelines as a reference), time to development of 1 new major protease inhibitor mutation, and time to loss of phenotypic susceptibility to 1 drug equivalent, as outlined below [8]. The following mutations were considered to be nucleoside reverse-transcriptase inhibitor (NRTI) resistance mutations: M41L, E44D, K65R, D67N, T69S (insertion complex), K70R, L74V, Y115F, V118I, Q151M, M184V/I, L210W, T215Y/F, and K219Q/E. A62V, V75I, F77L, and F116Y were not considered to be individual mutations because of their clinical significance only in the presence of Q151M [8]. The T69D mutation was excluded from this analysis because it is rare and of unclear clinical significance [8]. The following mutations were considered to be nonnucleoside reverse-transcriptase inhibitor (NNRTI) resistance mutations: L100I, K103N, V106A/M, V108I, Y181C/I, Y188C/L/H, G190A/S, P225H, M230L, and P236L. The following mutations were considered to be "major" protease inhibitor resistance mutations: D30N, L33F, M46I/L, G48V, I50V/L, V82A/F/T/S/L, I84V, and L90M. The following mutations were considered to be "minor" protease inhibitor mutations: L10I/R/V/F, K20M/R/I, L24I, V32I, L33I, M36I/L/V, I47V/A, F53L, I54V/L/M/A/T/S, A71V/T, G73S/A/C/T, V77I, and N88D/S. The L63P mutation was excluded because it is commonly observed in untreated individuals and is generally considered to be a common polymorphism [9].
It is unclear how to use emerging new antiretroviral drugs as they become available. One option is to preserve the use of a new drug until other potentially effective drugs become available. Therefore, we assessed the risk of deferring the use of 2 new protease inhibitors, tipranavir and darunavir. A revised tipranavir mutation score was calculated for subjects currently receiving protease inhibitors [10], and time to development of a 1-point increase in the revised score was assessed. The time to development of a new darunavir-associated mutation (V32I, L33F, I47V, I54L, or L89V) was also estimated [11].
"Total number of mutations" included all established NRTI, NNRTI, and protease inhibitor (major and minor) mutations [12], as well as the revised tipranavir [10] and preliminary darunavir mutations [11]; analyses excluding the unique tipranavir (V11L, I13V, K20V, E35G, K43T, Q58E, H69K, A71L, T74P, L76V, V82I/L, N83D, or L89V) and darunavir (L89V) mutations did not yield significantly different results (data not shown). Positions containing mixtures of wild-type and mutant residues were considered mutants.
Phenotypic resistance end points. All FDA-approved antiretroviral drugs were considered in this analysis, with the following exceptions: (1) stavudine was excluded because there is a high level of cross-resistance between zidovudine and stavudine [13], (2) emtricitabine was excluded because lamivudine and emtricitabine have identical resistance patterns [12], (3) nevirapine and delavirdine were excluded and efavirenz was selected as a representative NNRTI agent [14], and (4) amprenavir and its prodrug, fosamprenavir, were considered to be clinically equivalent [15]. Atazanavir, tipranavir, and darunavir were not included in this analysis because they were not included in the phenotypic assay at the time most of the testing was performed.
Each distinct antiretroviral drug (zidovudine, lamivudine, didanosine, abacavir, tenofovir, efavirenz, indinavir, nelfinavir, saquinavir, ritonavir, amprenavir, and lopinavir) was given a score that was based on phenotypic susceptibility (0, resistant; 0.5, partially susceptible; 1.0, fully susceptible). Scores were added to give a composite score for each time point. Time to loss of phenotypic susceptibility was defined as the loss of 1 fully effective drug equivalent (or 2 partially effective drugs).
Statistical methods. Kaplan Meier analyses were performed to determine time to development of genotypic and phenotypic end points. The log-rank test was used identify possible predictors of failure (baseline CD4+ T cell count, HIV RNA load, number of previous antiretroviral drugs, drug adherence, number of genotypic mutations, number of available antiretroviral drugs, phenotypic susceptibility score, and replication capacity) for the primary end points.
RESULTS
Baseline characteristics. A total of 106 individuals who were receiving antiretroviral drugs and had drug-resistant viremia were eligible for the study (table 1). Patients were observed for a median of 11.3 months (interquartile range [IQR], 7.4-21.0 months), with a median of 3 observations per subject (IQR, 2-5 observations). Baseline for this study was defined as the first visit in which subjects met the eligibility criteria. The median CD4+ T cell count and plasma HIV RNA levels at baseline were 292 cells/mm3 (IQR, 199-424 cells/mm3) and 3.74 log10 copies/mL (IQR, 3.30-4.34 log10 copies/mL), respectively. Subjects had previously received a median of 8 prior drugs (IQR, 5-10 prior drugs) . Most subjects (72%) were censored because of a change in therapy (change or discontinuation of antiretroviral regimen); the remaining subjects (28%) were administratively censored (end-of-study). An estimated 64% of the cohort had changed therapy by 12 months (data not shown).
Table 1. Baseline characteristics of 106 subjects receiving a partially suppressive treatment regimen
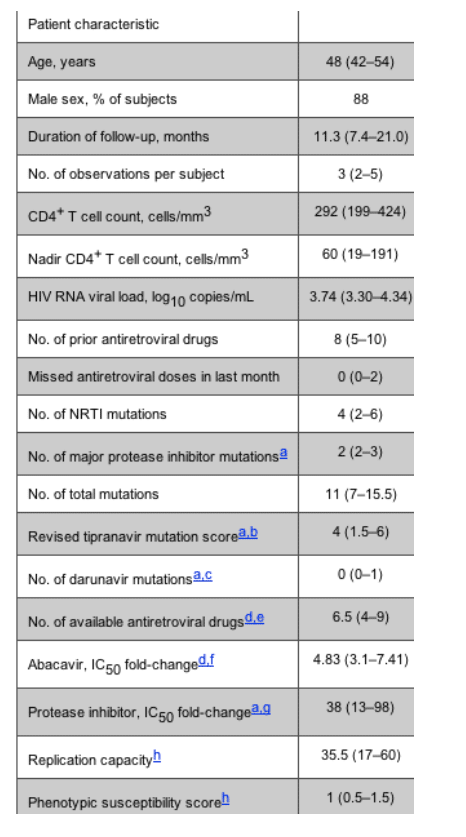
Genotypic end points. Using a Kaplan-Meier analysis, the estimated risk of developing 1 new nucleoside analogue mutation at 1 year was 23% (95% CI, 15%-34%). The risk of developing any new mutation at 1 year was 44% (95% CI, 33%-56%) (figure 1).
Figure 1. The time to development of at least 1 new nucleoside reverse-transcriptase inhibitor (NRTI) mutation, 1 new protease inhibitor (PI) mutation, and 1 new mutation is shown for 106 subjects who continued to receive a partially suppressive treatment regimen. Data for PI resistance mutations is shown only for the 71 subjects who were currently taking a PI.
Seventy-one patients were receiving a protease inhibitor during the study; the risk of developing a new major protease inhibitor mutation at 1 year for these patients was 18% (95% CI, 9%-34%) (figure 1). At 1 year, an estimated 26% (95% CI, 16%-40%) of protease inhibitor-treated patients experienced a 1-point increase in the revised tipranavir mutation score, and an estimated 8% of patients (95% CI, 3%-11%) developed a new darunavir-associated mutation. Because of their potential use in "salvage" therapy, the tipranavir and darunavir end points were compared among subjects with the fewest drug options (PSS = 0) and all others (PSS > 0). These results were not statistically significant (tipranavir, P = .24; darunavir, P = .37).
Patients who had fewer total mutations at baseline were more likely to develop a new nucleoside analogue mutation (above vs. below median, P = .01). Similarly, patients who had the fewest drug options at baseline (PSS = 0) were less likely to develop a new nucleoside analogue mutation, compared with those who had a PSS > 0 (P = .03). In addition, subjects with a higher replication capacity were more likely to develop a new nucleoside analogue mutation (above vs. below median, P = .03). Baseline CD4+ T cell count, HIV load, number of previous antiretroviral drugs, drug adherence, number of genotypic mutations, number of available antiretroviral drugs, phenotypic susceptibility score, and replication capacity were not consistently predictive of time to development of a new major protease inhibitor mutation.
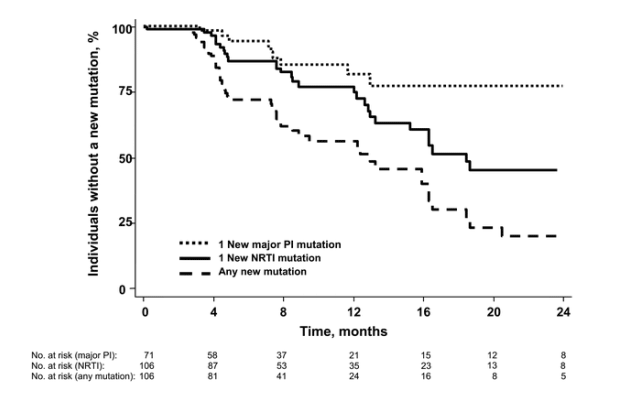
Phenotypic end points. The estimated risk of losing 1 fully suppressive drug equivalent (or 2 partially suppressive drugs) at 1 year was 30% (95% CI, 21%-43%; figure 2). There were no significant predictors of time to loss of 1 drug equivalent.
Figure 2. The time to loss of 1 fully suppressive drug (as defined phenotypically) is shown for 106 subjects who continued to receive a partially suppressive treatment regimen. Each distinct antiretroviral drug (see Methods) was assigned a score on the basis of phenotypic susceptibility (0, resistant; 0.5, partially susceptible; 1.0, fully susceptible). Time to loss of phenotypic susceptibility was defined as loss of 1 fully effective drug equivalent (or 2 partially effective drugs).
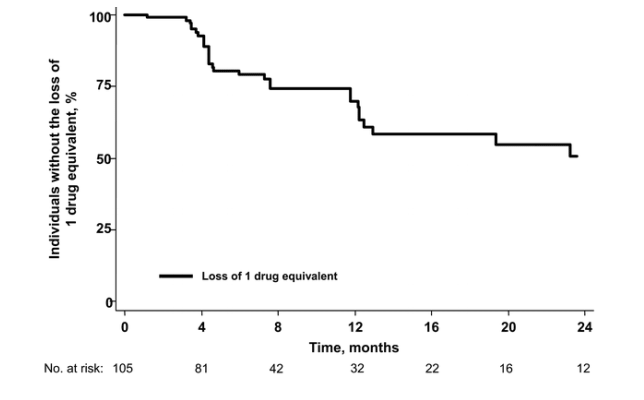
Loss of mutations and gain of drug options. A secondary analysis evaluating the converse end points of "loss" of measurable genotypic mutations and "gain" of drug options was also conducted. At 1 year, the estimated probability that an existing mutation would become undetectable by our population-based genotypic resistance assay was 14% (95% CI, 8%-24%) for nucleoside analogue mutations, 7% (95% CI, 3%-18%) for protease inhibitor mutations, and 32% (95% CI, 22%-44%) for any mutation (figure 3). Similarly, the estimated probability of "gaining" 1 fully effective drug equivalent at 1 year was 22% (95% CI, 14%-34%) (figure 4). Because established resistance mutations are known to persist indefinitely at low levels or in the "latent" reservoir [16-18], these apparent "losses" in resistance mutations and "gains" in phenotypic susceptibilities likely reflect fluctuations in the level of the resistance changes at or near the level of detectability.
Figure 3. The time to apparent loss of a measurable genotypic mutation is shown for 106 subjects who continued to receive a partially suppressive treatment regimen. Data for protease inhibitor (PI) resistance mutations is shown only for the 71 subjects who were currently receiving a PI. NRTI, nucleoside reverse-transcriptase inhibitor.
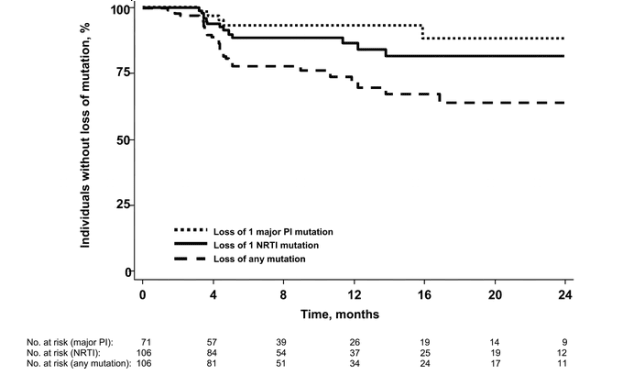
Development of genotypic mutations not known to be associated with drug resistance. Given the possibility that we were underestimating the rate of viral evolution by focusing our analysis on the known IAS-USA mutations [19], we also assessed the risk of developing any change from consensus sequence, excluding the known IAS-USA mutations (figure 5). At 1 year, the estimated risk of developing any reverse-transcriptase mutation, excluding the known NRTI mutations, was 88% (95% CI, 79%-94%). Similarly, in individuals currently receiving a protease inhibitor, the estimated risk of developing a protease mutation (excluding the known protease inhibitor mutations) was 64% (95% CI, 51%-77%). Interestingly, for individuals who were not receiving a protease inhibitor, the estimated risk of developing a protease mutation (excluding the known protease inhibitor mutations) was 67% (95% CI, 44%-87%), suggesting a substantial rate of viral "evolution," even without specific drug pressure.
DISCUSSION
In this study, we measured the risk of developing drug-resistance mutations over time in a cohort of treatment-experienced, HIV-infected patients who continued to receive (at the discretion of their primary care providers) a stable antiretroviral regimen despite incomplete viral suppression. An estimated 44% of patients developed at least 1 new drug resistance mutation over 1 year, whereas 30% lost the phenotypic equivalent of 1 susceptible drug at 1 year.
Similar rates of genotypic mutations have been reported by others [20]. However, to our knowledge, this is the first such longitudinal study to use both genotypic and phenotypic resistance testing performed at multiple, predetermined (rather than clinically determined) intervals. Kantor et al. [4] retrospectively identified 106 subjects receiving a stable regimen for whom resistance testing was performed as part of routine clinical care. After a median of 14 months, 75% of subjects developed a new resistance mutation. Napravnik et al. [5] used a similar study design and found that, among 98 subjects with persistent viremia who were receiving stable antiretroviral therapy, 60% acquired at least 1 new mutation during a median of 9.3 months. The higher rates of acquiring drug resistance mutations reported in these studies may be because of differences in disease stage and reason for genotyping. Eleven percent to 15% of subjects in these prior studies were receiving their first antiretroviral regimen [4, 5], compared with none in our study. Moreover, the study by Napravnik et al. [5] included subjects who did not have any resistance mutations at baseline. As suggested by our study and others, patients with the greatest risk of developing drug resistance mutations are those who have fewer mutations at baseline [4, 5].
The resistance cost of maintaining a stable regimen despite incomplete viral suppression argues for aggressive modification of therapy during virologic failure. However, for many patients, a fully suppressive option is not available, and any decision to switch therapy early risks the rapid loss of future drug options. Also, many-but not all-patients who continue to receive a partially suppressive regimen exhibit sustained immunologic and clinical benefits [21-23]. The mechanism for this benefit is likely to be multifactorial, and includes a combination of residual drug activity, decreased viral fitness, enhanced HIV-specific immune responses, and decreased T cell activation [24-26]. Future studies should be aimed at generating a prognostic algorithm that might allow a clinician to more accurately determine when the benefit of continuing a stable regimen outweighs the risk. With regard to predicting the risk of further evolution of drug resistance, we found that subjects who had fewer baseline mutations were more likely to develop at least 1 new nucleoside analogue mutation. Similarly, those who were not currently receiving any "active" drugs were less likely to develop a new nucleoside analogue mutation. These data, as well as those of others [4, 5], indicate that patients with less resistance at baseline have the highest risk for losing future drug options.
Several limitations of our study deserve comment. First, because we only included subjects who had been receiving a stable regimen for at least 120 days, this analysis may have selected for subjects who were clinically stable. However, an estimated 19%, 46%, and 64% of our subjects had changed therapy by 4, 8, and 12 months, respectively, suggesting that we enrolled a dynamic cohort and did not select for patients who were clinically stable. Second, application of our findings may be limited by the fact that >1 regimen had failed for all of our subjects. Presumably, the rate of accumulation of drug resistance would be far greater among patients for whom their initial regimen failed. Also, studying resistance changes over time can only be done with patients who continue to receive a stable regimen. Therefore, our data are generalizable to patients who are either clinically stable or who have essentially no other therapeutic options. Third, the phenotypic analysis examining the rate of loss of 1 drug equivalent may underestimate the overall loss of drugs, because some subjects may have lost >1 drug. A similar concern applies to our genotypic data. Fourth, we were only able to estimate the time to development of tipranavir and darunavir resistance on the basis of our evolving understanding of the resistance mutations associated with these drugs [10, 11]. A more precise manner to address the rate of loss of activity for these protease inhibitors would likely require phenotypic data [27].
Although we observed a moderate rate of acquisition of new mutations, we also observed a significant rate of "losing" drug resistance mutations (or of "gaining" a phenotypically effective drug). We do not believe that this apparent "loss" of drug resistance mutations actually reflects a true loss of resistance, because it is well established that once resistance develops in an individual, resistance persists indefinitely [16-18]. Rather, we believe that both of these changes likely reflect oscillations of minority variants that exist at or near the level of detection [28, 29]. Alternatively, these changes may reflect novel evolutionary pathways that the virus is taking in response to treatment. Although the mechanism for these changes could not be addressed, it is apparent from our data that a single resistance test may underestimate the true level of drug resistance.
The risk of losing future drug options is likely to be the major risk associated with maintaining a partially suppressive regimen. Although we observed a significant risk of viral evolution overall, the risk of accumulating genotypic resistance to tipranavir and darunavir was relatively low. Carefully defining the risk of losing specific drugs that are commonly used for "salvage" therapy may be a more precise manner in which to balance the risks and benefits of not switching therapy in patients with limited therapeutic options. Ultimately, the clinical scenario will require balancing the potential of viral evolution with the benefit of preserving antiretroviral drugs until others are available to administer in combination. In the setting of relative clinical stability, it may be prudent to wait to switch therapy until at least 2 new effective antiretroviral agents are available.
|
|
| |
| |
|
|
|