| |
New Highly Sensitive HIV test may predict drug resistance at 0.1-.01%
|
| |
| |
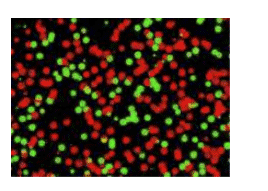
The test identifies which drug-resistant strains of HIV are harbored in a patient's bloodstream. Viruses that have evolved resistance to HIV drugs are tagged to appear green, while those that...
"......detection of resistance mutations, especially those present as minor populations, is critical to understanding mechanisms of multiple-drug resistance, to assisting the design of the best treatment regimens and to predicting treatment outcomes....We developed a highly sensitive parallel allele-specific sequencing (PASS) assay to simultaneously analyze a large number of viral genomes and detect minor drug-resistant populations at about 0.1-0.01% levels. Using this assay on samples from individuals infected with human immunodeficiency viruses (HIV), we successfully detected and quantified minor populations of drug-resistant viruses and performed linkage analysis of multiple-drug resistance mutations. This assay may serve as a useful tool to study drug resistance in HIV and other infectious agents...." See article text below
press release date: 7-Jan-2007
Contact: Marla Vacek Broadfoot
marla.broadfoot@duke.edu
919-660-1306
Duke University Medical Center
DURHAM, N.C. -- Researchers at Duke University Medical Center have developed a highly sensitive test for identifying which drug-resistant strains of HIV are harbored in a patient's bloodstream.
The test may provide physicians with a tool to guide patient treatment by predicting if a patient is likely to become resistant to a particular HIV drug, said one of its developers, Feng Gao, M.D., associate professor of medicine. Drug resistance is one of the most common reasons why therapy for HIV, the virus that causes AIDS, fails.
The test, which detects genetic changes, or mutations, in HIV, also may help scientists understand how the constantly evolving virus develops drug resistance, Gao said. He said such knowledge ultimately may result in the development of new treatments designed to evade resistance.
The findings will appear online on Sunday, Jan. 7, 2007, in the journal Nature Methods, as well as in the journal's February 2007 print edition. The work was supported by the National Institutes of Health and the Duke Center for AIDS Research.
Duke has filed for a provisional patent on the technology, and the researchers are considering ways to establish a new company to pursue its development or to license the technology to an existing company, Gao said.
Because HIV genes mutate so easily and the virus reproduces so rapidly, most people who are infected have many different forms of the virus in their bodies. In some cases, mutated strains take on new properties that make them more resistant to the drugs used in antiretroviral therapy, the primary means of treatment for HIV infection.
During antiretroviral therapy that does not fully suppress the virus, a strain that develops drug resistance will grow more quickly than strains lacking such resistance, and the resistant strain will replicate to become the most prominent virus in the person's body.
"The viral populations found in the blood of one patient can be very different from the populations present in another," Gao said. "Which resistant viruses are at hand can have important implications for the successful treatment of that patient."
More than 20 drugs currently are available for treating HIV infection. All but one of the drugs target two of the genes that serve as blueprints for vital protein components of HIV: reverse transcriptase and protease.
The Duke test examines the genes of HIV strains for mutations at certain positions that are known to be linked to drug resistance. For example, a change at a specific spot along the genetic code -- position 46 -- of the protease gene results in resistance to the drug indinavir.
To assess the test, the researchers analyzed blood samples from three different groups of HIV patients: those who had never received antiretroviral treatment, those who had received treatment but were not currently being treated and those who were receiving treatment but the treatment was not completely successful.
After processing the blood samples and isolating the genetic material in each of them, the researchers added tiny fluorescent tags designed to stick to HIV genes in particular ways. Tags designed to stick to mutated gene locations known to produce drug resistance were labeled to appear green, while tags designed to stick to the same gene locations but where the genes had not mutated were labeled to appear red.
The researchers used a sophisticated computer program to count the number of molecules with green or red fluorescent tags in each sample. The test proved sensitive enough to detect a single mutated virus out of 10,000 nonmutated viruses in the patient samples, Gao said.
"This level of sensitivity makes the assay about 1,000 times more sensitive than the most widely used assays on the market for detecting drug-resistant HIV viruses" Gao said. "Thus, the assay may permit more accurate prediction of treatment outcomes."
The test also can detect when a virus molecule has more than one mutation, a capability that no commercially available test has achieved, Gao said. This capability may prove critical for detecting HIV strains that have become resistant to multiple drugs, a condition that occurs often as many patients are treated with many drugs at the same time.
The test may find broader medical application as well, Gao said. He said it has the potential to detect mutations that confer drug resistance in infectious agents that cause other diseases besides HIV, such as hepatitis B, hepatitis C and tuberculosis.
Richard Shafer of the HIV Drug Resistance Database at Stanford University in California, US, agrees there is a need for improved detection of mutant strains. "The stakes are high when a physician has to choose therapy for a patient with a drug-resistant virus," he explains.
"The correct combination of drugs can halt virus replication and disease progression in its tracks. An incorrect combination of drugs not only enables the virus to continue to replicate but also makes a patient's virus resistant to even more drugs."
Approximately 10% to 20% of HIV-infected people in the US and Europe carry drug-resistant strains and that percentage appears to be increasing. Shafer notes that early detection might ultimately curb the spread of drug-resistant HIV strains by reducing their presencein the population.
Duke has filed for a provisional patent on the technology, and researchers there are exploring whether they might license it to a company for development.
However, Shafer says there are major challenges that this and other methods must overcome before being widely adopted. For example, some of the mutant virus might not be detected while it hides inside cells: "Many of the deadly minor drug-resistant viruses are not in plasma but reside instead in a semi-latent form in cells, which are not present at large numbers in blood samples," he explains.
Journal: Nature Methods (DOI: 10.1038/nmeth995)
Detection of minor drug-resistant populations by parallel allele-specific sequencing
Brief Communication
Published online: 7 January 2007; | doi:10.1038/nmeth995
Fangping Cai1, 5, Haifeng Chen1, 5, Charles B Hicks2, John A Bartlett2, Jun Zhu3, 4 & Feng Gao1
1 Duke Human Vaccine Institute, Department of Medicine, Duke University Medical Center, Durham, North Carolina 27710, USA.
2 Division of Infectious disease, Department of Medicine, Duke University Medical Center, Durham, North Carolina 27710, USA.
3 Institute for Genome Sciences and Policy, Duke University Medical Center, Durham, North Carolina 27710, USA.
4 Department of Cell Biology, Duke University Medical Center, Durham, North Carolina 27710, USA.
We developed a highly sensitive parallel allele-specific sequencing (PASS) assay to simultaneously analyze a large number of viral genomes and detect minor drug-resistant populations at 0.1-0.01% levels. Using this assay on samples from individuals infected with human immunodeficiency viruses (HIV), we successfully detected and quantified minor populations of drug-resistant viruses and performed linkage analysis of multiple-drug resistance mutations. This assay may serve as a useful tool to study drug resistance in HIV and other infectious agents.
Highly active antiretroviral therapy (HAART) is the primary approach to treat HIV infection. However, accumulation of drug resistance mutations in viral genomes can lead to treatment failure1. Therefore, detection of resistance mutations, especially those present as minor populations, is critical to understanding mechanisms of multiple-drug resistance, to assisting the design of the best treatment regimens and to predicting treatment outcomes2. Pre-existing minor populations of drug-resistant viruses may outgrow the wild-type population and become predominant when drug selection pressure is present.
To detect such minor resistant populations, we developed the PASS assay to simultaneously analyze a large number of viral genomes by applying the polony technique3, 4. Using this assay, we amplified a 1.1 kb pol gene fragment containing sites of all major resistance mutations in reverse transcriptase and protease. Because one acrydited primer becomes immobilized by covalently incorporating into polyacrylamide gels during the polymerizaton, the PCR products accumulate around individual DNA templates and form distinct spots (polonies) at the amplification sites (Supplementary Fig. 1 and Supplementary Methods online). After amplification, the solid-phase negative DNA strands hybridize to complementary sequencing primers whose 3' end is juxtaposed to the site where a single-base mutation confers resistance. After single-base extension of this primer in the presence of nucleotides labeled with different fluorophores (Supplementary Fig. 1), imaging with a microarray scanner can be used to distinguish wild-type and mutant populations (Supplementary Fig. 2 online).
To determine the sensitivity of the PASS assay, we spiked wild-type plasmid molecules (plasmid WEAU-wt) with mutant molecules (plasmid WEAU-E44D) at various ratios (1:1, 1:10, 1:100 and 1:1,000), and probed the mixtures with the E44D primer. We easily detected mutations present at 0.1% or higher (Fig. 1a). When we increased the total viral genome input to 10,000 at the same 1:1,000 mutant/wild-type ratio, we also readily detected resistance mutations, although a proportion of wild-type molecules were fused to one another and could not be accurately counted (Supplementary Fig. 2). Therefore, the assay sensitivity for detection of minor resistance mutations may be as high as about 0.01%. Linear regression analysis revealed a good linear correlation (R2 = 0.9945) between detected and expected mutant/wild-type ratios (Supplementary Fig. 2).
We detected 22 primary mutations on individual molecules through multiple single-base extension reactions, suggesting that the PASS assay can be used to detect most primary mutations and potentially other secondary mutations for linkage analysis when more sequencing cycles are performed (Supplementary Fig. 3 online). We also tested 27 viral genomes from many subtypes and recombinant forms (2 As, 8 Bs, 5 Cs, 3 Ds, 1 F, 2 Gs, 1 H, 2 CRF01_AEs, 1 CRF04_cpx and 2 other recombinants). All tested subtypes and recombinants were comparably amplified, suggesting that the PASS assay can be used for most genetic subtypes and recombinants (Supplementary Fig. 4 online). The sensitivity of the assay for detection of both wild-type and mutant populations was about 6 copies of viral genomes. Linear regression analysis showed a good linear detection correlation (R2 = 0.9958) between 10 and 10,000 copies of viral genomes (Supplementary Fig. 5 online). We detected no unexpected mutant bases when more than 200,000 wild-type molecules were analyzed, suggesting that the mutation rate introduced by Taq polymerase was lower than 5 10-6 under the present experimental conditions. Therefore, low frequency mutations detected by PASS will not likely represent PCR errors.
To detect different resistance mutations in each molecule present in a viral population, we generated three single drug-resistance clones (WEAU-E44D, WEAU-L90M and WEAU-M184V) and mixed them with the wild-type clone (WEAU-wt) at a 1:1:1:1 ratio. We then sequentially sequenced the polonies with E44D, M184V and L90M primers. As expected, about 25% of the polonies contained the resistance mutation and the other 75% of polonies were wild type in each reaction (Fig. 1b-d). When polonies were analyzed from all three images, the wild-type clones showed as a green-green-red pattern, whereas E44D clones showed as red-green-red, M184V as green-red-red and L90M as green-green-green. This result demonstrates that the PASS assay can accurately determine the presence of different resistance mutations in each viral genome for linkage analysis when a large number of viral genomes are analyzed in parallel.
To detect resistance populations in samples from HIV-infected patients using the PASS assay, we analyzed 13 plasma samples from three different patient groups: patients who were never treated before (naive), patients previously treated but not currently on antiretroviral therapy (previously treated), and patients failing a current treatment regimen (treatment failure). The Institutional Review Board exemption for working with pre-existing human specimens in this study was approved by the Duke University Health System Institutional Review Board. We first determined the frequency of M184V mutation and detected no mutations in treatment-naive patients, minor drug-resistant populations (<2%) in two of six previously treated patients, and major drug-resistant populations in treatment-failure patients (36-100%; Supplementary Fig. 6 and Supplementary Table 1 online).
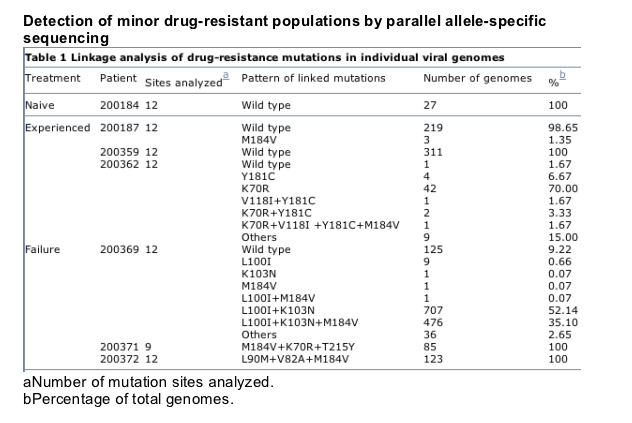
To determine whether other resistance mutations were present among studied viral populations, we analyzed 12 potential resistance mutation sites in seven patient samples representing the three groups (Table 1). We detected additional resistance mutations in one previously treated patient and in all three treatment-failure patients. We detected L90M and V82A mutations in patient 200372, and K70R and T215Y mutations in patient 200371 (Table 1 and Supplementary Fig. 6). We found no wild-type viruses in either patient. In two other patients, we detected both resistance-associated and wild-type sequences at multiple resistance-mutation sites in the viral genomes (Fig. 2). This allowed us to perform detailed linkage analysis. In treatment-failure patient 200369, the L100I mutation was present in 90.32% of the viruses and the K103N mutation was present in 89.64% of the viruses. The 36.26% M184V mutant viruses were not detected in the standard genotypic assay but were readily detected in the PASS assay (Fig. 2). Linkage analysis showed that more than half of the viruses (52.14%) carried both L100I and K103N mutations, and 35.10% of the viruses had three resistance mutations (L100I, K103M and M184V). Other viruses containing single or dual resistance mutations were present as minor populations (L100I, 0.66%; K103N, 0.07%; M184V, 0.07%; and M100I/M184V, 0.07%; Table 1). The previously treated patient 200362 also showed a similar resistance-mutation linkage profile. Although the vast majority of viruses contained one or more resistance mutations, both patients still harbored minor wild-type virus populations (1.67% and 9.22%).
Consistent improvement in HIV treatment outcomes as a consequence of genotypic or phenotypic testing among patients failing antiretroviral treatment has not been confirmed5. This may partially be due to the low sensitivity (presence of >20% mutant viruses) of the assays for detection of minor drug-resistance populations in such patients6, 7. Point mutation assays offer substantially greater sensitivity than either genotypic or phenotypic assays7. As drug-resistance mutations detected by these methods are population-based, none of them allow linkage analysis. Instead, clonal sequencing has been used to study minor resistance populations and linkage analysis of resistance mutations on each viral genome8. Although it takes a few days to analyze all primary mutations using the PASS assay, this approach is substantially more efficient, faster and more sensitive for detection of minor resistance populations than is clonal sequencing. Although the PASS assay takes longer to complete than standard genotypic assays, it offers better sensitivity for detection of minor resistance mutations and for linkage analysis. While the clinical significance of minor resistance viral populations has not been fully determined, it is likely that such populations affect subsequent treatment outcomes considerably2, 9, 10. Because the PASS method is up to 1,000 times more sensitive than either genotypic or phenotypic assay (0.01% versus 20%) for detection of minor resistance viral populations, it may permit more accurate prediction of treatment outcomes. The PASS assay permits a detailed linkage analysis of multiple mutations, allowing study of the impact of different combinations of mutations existing as minor and major viral populations (Table 1). In addition to these clinical improvements, technical advantages of the PASS assay are notable. For example, in the PASS procedure viral cDNA molecules are directly embedded into polyacrylamide gel, and PCR amplification is carried out at a single-molecule level. Therefore, artifact sequences that are generated through recombination or resampling during conventional PCR are eliminated11, 12.
Here we describe a PASS assay that can detect minor resistance populations in HIV-infected individuals with high sensitivity, specificity and throughput. The approach is an application of the polony technique, which has been used for profiling combinatorial alternative pre-mRNA splicing, parallel detection of single-base mutation and parallel sequencing4, 13, 14, 15. The new technology may also potentially be used to detect HIV escape mutations in antigen determinants targeted by T and B cells. Its further use and clinical application is the subject of ongoing research.
|
|
| |
| |
|
|
|