| |
Rapid and Durable Antiretroviral Effect of the HIV-1 Integrase Inhibitor Raltegravir as Part of Combination Therapy in Treatment-Naive Patients With HIV-1 Infection: Results of a 48-Week Controlled Study
[Rapid Communication]
|
| |
| |
JAIDS Journal of Acquired Immune Deficiency Syndromes:Volume 46(2)1 October 2007pp 125-133
Markowitz, Martin MD; Nguyen, Bach-Yen MD; Gotuzzo, Eduardo MD; Mendo, Fernando MD; Ratanasuwan, Winai MD; Kovacs, Colin MD; Prada, Guillermo MD#; Morales-Ramirez, Javier O MD; Crumpacker, Clyde S MD; Isaacs, Robin D MD; Gilde, Lucinda R BS; Wan, Hong MS; Miller, Michael D PhD; Wenning, Larissa A PhD; Teppler, Hedy MD; the Protocol 004 Part II Study Team
From the Aaron Diamond AIDS Research Center, Rockefeller University, New York, NY; Merck Research Laboratories, West Point, PA; Hospital Nacionale Cayetano Heredia, Lima, Peru; Hospital Nacionale Edgardo Rebagliati, Lima, Peru; Siriraj Hospital, Bangkok, Thailand; Canadian Immunodeficiency Research Collaborative, Toronto, Ontario, Canada; #Fundacion Santafe de Bogota University Hospital, Bogota, Colombia; Clinical Research Puerto Rico, Inc., San Juan, PR; and Beth Israel Deaconess Medical Center, Boston, MA.
"....Raltegravir was more lipid neutral than efavirenz based on levels of total cholesterol, LDL-C, and triglycerides.....all doses of raltegravir in combination with tenofovir and lamivudine displayed potent antiretroviral activity comparable to that of efavirenz in combination with tenofovir and lamivudine, a current standard of care for treatment-naive patients.... All doses of raltegravir, up to 600 mg twice daily, in combination with tenofovir and lamivudine were generally well tolerated.... the percent of subjects remaining below 400 and 50 HIV-1 RNA copies/mL, respectively, at 48 weeks was maintained across all raltegravir treatment groups when compared with responses at week 24 and was comparable to that observed in the efavirenz treatment group..... rate at which HIV-1 RNA levels fell to undetectable levels was more rapid for all doses of raltegravir when compared with the efavirenz group .....Virologic failure was uncommon and observed in only 5 patients (3%) receiving raltegravir and 1 patient (3%) receiving efavirenz. In the patients receiving raltegravir, 4 had resistance-conferring mutations in the RT region: K65R in 1 and M184V and/or M184I in all 4....
....In summary, this report describes the longest duration of treatment (48 weeks) with the integrase inhibitor raltegravir in treatment-naive patients infected with HIV-1. The results of this study demonstrate that raltegravir in combination with tenofovir and lamivudine has rapid, potent, and durable antiretroviral efficacy and a favorable safety profile compared with efavirenz plus tenofovir and lamivudine. Given this profile and these data, the use of raltegravir in combination with nucleoside reverse transcriptase inhibitors (NRTIs) warrants further evaluation as a potential first-line regimen."
Abstract
Background: Raltegravir is an HIV-1 integrase strand-transfer inhibitor with potent in vitro activity. This study explored the antiretroviral activity and safety of raltegravir in treatment-naive patients with plasma HIV-1 RNA levels ≥5000 copies/mL and CD4+ T-cell counts ≥100 cells/mm3.
Methods: Multicenter, double-blind, randomized, controlled study of raltegravir at doses of 100, 200, 400, and 600 mg twice daily versus efavirenz at a dose of 600 mg/d, all in combination with tenofovir at a dose of 300 mg/d and lamivudine at a dose of 300 mg/d (clinicaltrials.gov identifier: NCT00100048).
Results: In the 198 patients treated (160 on raltegravir and 38 on efavirenz), the mean HIV-1 RNA level ranged from 4.6 to 4.8 log10 copies/mL at baseline. At weeks 2, 4, and 8, the proportion of patients achieving an HIV-1 RNA level <50 copies/mL was greater in each of the raltegravir treatment groups than in the efavirenz group. By week 24, all treatment groups appeared similar, with plasma HIV-1 RNA levels <400 copies/mL in 85% to 98% of patients and <50 copies/mL in 85% to 95% of patients. These reductions were maintained through week 48 in 85% to 98% of patients and in 83% to 88% of patients, respectively. Five (3%) patients on raltegravir and 1 (3%) on efavirenz experienced virologic failure before week 48. Drug-related clinical adverse events were less common with raltegravir than with efavirenz. After 24 and 48 weeks of treatment, raltegravir did not result in increased serum levels of total cholesterol, low-density lipoprotein cholesterol, or triglycerides.
Conclusions: Raltegravir at all doses studied was generally well tolerated in combination with tenofovir and lamivudine. Raltegravir exhibited potent and durable antiretroviral activity similar to that of efavirenz at 24 and 48 weeks but achieved HIV-1 RNA levels below detection at a more rapid rate.
Despite the success of highly active antiretroviral therapy in reducing HIV-1-related mortality and morbidity, current treatment regimens may be limited by the emergence of drug resistance and short- and long-term toxicities. Up to 88% of patients with detectable viremia despite treatment with antiretroviral therapy harbor virus with reduced susceptibility to at least 1 therapeutic class, with more than 50% resistant to multiple classes.1 Furthermore, transmission of drug-resistant HIV-1 has been well documented: the prevalence of drug-resistant HIV-1 mutations in untreated individuals ranges from 8% to 27% and includes resistance to more than 1 drug class in up to 13%.2-7 Transmitted drug-resistant mutations can persist for years8-10 and may be associated with a less favorable response to antiretroviral therapy.5,6,11 Clearly, new drugs directed against novel targets are needed for treatment-naive and treatment-experienced patients. Raltegravir (formerly known as MK-0518) is a strand-transfer inhibitor of HIV-1 integrase, which is essential for viral replication and therefore provides a unique and specific target for antiretroviral drug development.12
In treatment-experienced patients with multidrug-resistant HIV, raltegravir with optimized background therapy (OBT) has been shown to be generally well tolerated, with a safety profile comparable to that of placebo plus OBT, and to provide significantly better viral suppression than placebo plus OBT at 16 and 24 weeks.13-15 In treatment-naive patients, 10 days of monotherapy with raltegravir (100, 200, 400, or 600 mg twice daily) resulted in an approximately 2.0 log10 reduction in plasma HIV-1 RNA levels.16 Here, we present the results of the second part of that study, which evaluated the safety and efficacy of raltegravir after 48 weeks of combination therapy with tenofovir and lamivudine in treatment-naive HIV-1-infected patients. The control regimen selected for this study, efavirenz plus tenofovir and lamivudine, is a potent and durable antiretroviral treatment regimen17 recommended as initial therapy in current Department of Health and Human Services (DHHS) guidelines.18 Preliminary data from drug-drug interaction studies demonstrated satisfactory raltegravir levels and tolerability when used in combination with tenofovir and suggested no need for dosage adjustments.19
RESULTS
Patient Characteristics
A total of 201 patients were enrolled in the study (Fig. 1); 198 patients received study therapy and were included in the analyses. Baseline characteristics were balanced across treatment groups (Table 1). Plasma HIV-1 RNA levels were >50,000 copies/mL at screening in 55% of patients and >100,000 copies/mL at baseline in 34%. The mean HIV-1 RNA level at baseline ranged from 4.6 to 4.8 log10 copies/mL. The mean CD4+ T-cell count at baseline ranged from 271 to 338 cells/mm3. A total of 185 patients completed at least 48 weeks of study therapy. Based on medication diaries, at least 90% compliance (defined as taking at least 1 tablet of study medication per day) with the treatment regimen was reported by 98% of patients.
Efficacy
At week 24, plasma HIV-1 RNA levels were reduced to <400 copies/mL in 82% to 100% of cohort II and in 85% to 98% of cohorts I and II combined. Other outcomes (HIV-1 RNA level <50 copies/mL and change from baseline in CD4+ T-cells) at weeks 24 and 48 were also comparable for these groups. Therefore, the following results are based on cohorts I and II combined.
Raltegravir at all doses, in combination with tenofovir and lamivudine, resulted in a rapid and sustained reduction in HIV-1 RNA levels, with at least 90% of patients reaching below 400 copies/mL by week 4 (Fig. 2A). Similarly, rapid and sustained responses also were observed with all raltegravir doses when assessing the proportion of patients with a plasma HIV-1 RNA level <50 copies/mL (see Fig. 2B). At weeks 2, 4, and 8, HIV-1 RNA levels <50 copies/mL were achieved by more patients in each of the raltegravir groups than in the efavirenz group (lower bound of 95% CI >0). These differences diminished with time, and by week 24, all treatment groups appeared similar, with the plasma HIV-1 RNA level reduced to <50 copies/mL in 85% to 95% of patients (Table 2). These reductions in viral load were maintained through week 48 in 85% to 98% and 83% to 88% of patients, respectively. In a time-to-event analysis (see Fig. 2D), patients receiving raltegravir at any dose achieved an HIV-1 RNA level <50 copies/mL earlier than patients receiving efavirenz (log-rank test, P < 0.05).
The mean change in CD4+ T-cell count ranged from 70 to 104 cells/mm3 across all treatment groups at week 2 and continued to rise through week 32 (see Fig. 2C). The average increase was comparable across treatment groups at weeks 24 and 48 (see Table 2).
All doses of raltegravir showed potent antiretroviral efficacy in patients with an HIV-1 RNA level >50,000 copies/mL at screening and in patients with a lower viral load at screening (data not shown). Similarly, patients with a baseline HIV-1 RNA level >100,000 copies/mL achieved viral load reductions comparable to those observed in patients with <100,000 copies/mL at baseline (Table 3).
Virologic failure occurred in 5 (3%) of 160 patients receiving the raltegravir combination regimens and in 1 (3%) of 38 patients receiving the efavirenz combination regimen. Plasma-derived HIV-1 RNA isolated from 2 of the raltegravir patients at virologic failure harbored the N155H amino acid substitution in the integrase region, which was not present before treatment. In 1 of these patients, whose virus was genotyped after 24 weeks of therapy, the integrase gene contained 3 additional mutations (V151I, G163G/R, and D232D/N) relative to the baseline sequence and the reverse transcriptase (RT) gene contained K65R and a mixed population at the 184 codon (M/I/V). Nevertheless, this patient remained on study and had a >1.0-log reduction in plasma HIV-1 RNA at week 48. In the second patient with N155H, whose virus was genotyped after 8 weeks of therapy, the RT gene also had M184I/V; this patient discontinued the study after virologic relapse at week 8. Two of the raltegravir patients had resistance-conferring mutations detected in the RT region only (M184I/V), and 1 harbored only virus with no known resistance mutations at failure. The patient in the efavirenz group who had virologic relapse displayed genotypic and phenotypic resistance to efavirenz, tenofovir, and lamivudine; specific mutations were K65R and G190E in the RT region and S230N in the integrase region (a common polymorphism not thought to affect the activity of integrase inhibitors).
Pharmacokinetics
To characterize the effects of tenofovir and lamivudine on raltegravir, PK parameter values (area under the curve from 0 to 12 hours [AUC0-12h], maximum concentration [Cmax], and concentration at 12 hours [C12h]) were compared in patients who participated in both the monotherapy and combination therapy phases. Consistent across doses, raltegravir PK parameter values were, in general, slightly higher during combination therapy compared with monotherapy. The geometric mean AUC0-12h, Cmax, and C12h ratios (90% CIs) across all doses were 1.41 (1.11 to 1.79), 1.33 (0.96 to 1.85), and 1.42 (0.89 to 2.28), respectively. Steady-state PK parameter values for lamivudine were similar across treatment groups in cohort I patients during the combination therapy phase. The geometric mean AUC0-24h, Cmax, and C24hr ratios (90% CIs) across all doses were 1.40 (0.95 to 2.07), 1.20 (0.98 to 1.47), and 1.11 (0.27 to 4.61), respectively.
Safety
Most adverse events were graded mild to moderate, and there was no association between the frequency of adverse events and the dose of raltegravir (Table 4). The incidence of serious adverse events was similar in patients receiving the raltegravir combination regimens (6% overall) and those receiving the efavirenz combination regimen (5%). None of the serious adverse events were considered to be drug related or led to treatment discontinuation. Drug-related clinical adverse events were reported by fewer patients in the raltegravir treatment groups than in the efavirenz group (P = 0.04 for 100, 400, and 600 mg; P = 0.07 for 200 mg). Treatment-related adverse events with a >10% incidence included nausea in patients receiving raltegravir and dizziness, headache, abnormal dreams, nausea, diarrhea, insomnia, and nightmares in patients receiving efavirenz. Neuropsychiatric symptoms (abnormal dreams, depression, nightmares, and suicidal ideation) were less common in the raltegravir groups than in the efavirenz group during the first 8 weeks of therapy and also over the entire 48 weeks (Table 5), primarily because of differences in reports of abnormal dreams (7% vs. 21%) and nightmares (0% vs. 11%).
Few grade 3 and 4 laboratory abnormalities were observed during this study. In patients receiving raltegravir, these included decreased absolute neutrophil count (n = 1) and increases in aspartate aminotransferase (n = 3), alanine aminotransferase (n = 1), alkaline phosphatase (n = 1), pancreatic amylase (n = 3), and lipase (n = 1). In patients receiving efavirenz, grade 3 and 4 laboratory abnormalities included increases in low-density lipoprotein cholesterol (LDL-C; n = 1), triglycerides (n = 2), aspartate aminotransferase (n = 1), and alanine aminotransferase (n = 2). One patient in the raltegravir 600-mg group who was receiving concomitant isoniazid was prematurely discontinued because of increased aspartate aminotransferase (grade 4).
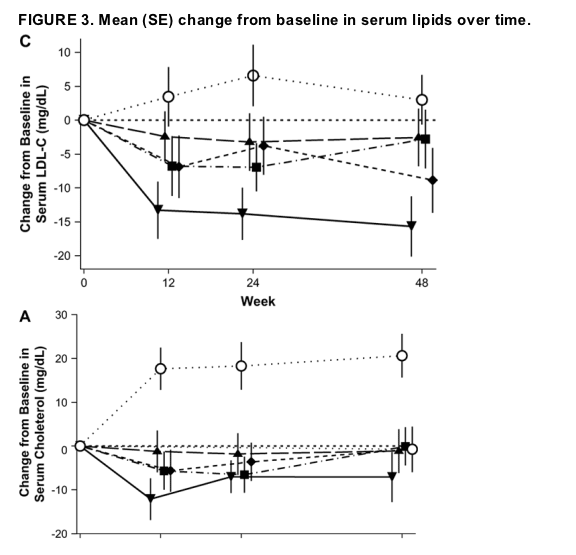
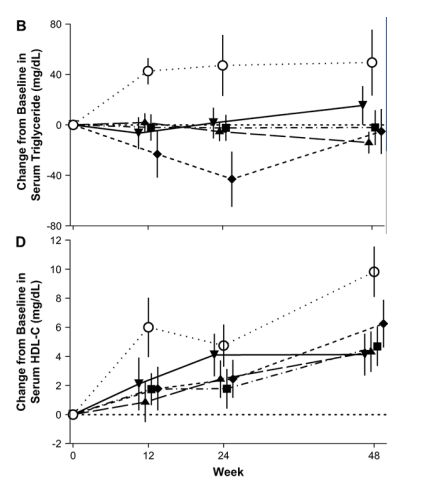
Baseline serum levels of total cholesterol, LDL-C, high-density lipoprotein cholesterol (HDL-C), and triglycerides were similar across treatment groups. At weeks 24 and 48, total cholesterol, LDL-C, and triglycerides were relatively unchanged in the raltegravir groups (Fig. 3) but were increased in the efavirenz group (P < 0.001, P = 0.016, and P = 0.07, respectively, vs. all raltegravir doses combined, week 48). HDL-C levels were increased in all treatment groups, but smaller increases were observed with raltegravir than with efavirenz (P = 0.01 at week 48). The total/HDL cholesterol ratio was decreased in all treatment groups at week 48; for all raltegravir doses combined, the mean change (from baseline value of 4.59) was -0.59; for the efavirenz group, the mean change (from baseline value of 4.72) was -0.47 (P = 0.52). Additionally, there were no clinically meaningful differences in the changes from baseline in body measurements among treatment groups (data not shown).
DISCUSSION
In this controlled randomized study, all doses of raltegravir in combination with tenofovir and lamivudine displayed potent antiretroviral activity comparable to that of efavirenz in combination with tenofovir and lamivudine, a current standard of care for treatment-naive patients. The rate at which HIV-1 RNA levels fell to undetectable levels was more rapid for all doses of raltegravir when compared with the efavirenz group, however. The rate at which HIV-1 RNA levels decrease after treatment initiation is biphasic, with a rapid first phase occurring over days, followed by a slower second phase lasting weeks. The first phase, estimated to be of 4 to 7 days in duration, reflects the short lifespan, approximately 1.4 days on average, of the activated, virus-producing, CD4+ T cell, whereas the second phase mirrors multiple processes, including the decay of longer-lived cells producing HIV-1 virions and the release of viral particles from follicular dendritic cells and particle production from cells latently infected and activated to produce virions.21,22 Although the design of this study does not allow us to discern whether the observed effect is limited to the first phase of decay, the second, or possibly both, we can attribute the accelerated HIV-1 RNA decay to more rapid and complete distribution of drug to sites and cells capable of producing HIV-1 particles or perhaps to greater inherent antiviral activity once delivered to the site of potential virus replication. Although we could not rule out an effect of raltegravir on the decay rate of virus-producing cells, we are hard-pressed to propose a mechanism for such. Interventional studies with frequent assessments of HIV-1 RNA levels in patients treated with optimally dosed raltegravir and efavirenz-based combination therapy are likely to shed more light in understanding the HIV-1 RNA decay differences seen in this study.
Not only is initial suppression of plasma viremia critical for achieving desired treatment responses, but durability of that response is essential because it indicates tolerability and the lack of emergence of drug-resistant virus. In the current study, the percent of subjects remaining below 400 and 50 HIV-1 RNA copies/mL, respectively, at 48 weeks was maintained across all raltegravir treatment groups when compared with responses at week 24 and was comparable to that observed in the efavirenz treatment group.
Virologic failure was uncommon and observed in only 5 patients (3%) receiving raltegravir and 1 patient (3%) receiving efavirenz. In the patients receiving raltegravir, 4 had resistance-conferring mutations in the RT region: K65R in 1 and M184V and/or M184I in all 4. Two of these patients had treatment-associated amino acid substitutions in the integrase region. Although limited in number, this is the first description of the emergence of resistance to the hydroxypyrimidinone carboxamide class of integrase inhibitors in humans. In both cases, the N155H substitution was identified alone or in combination with other integrase mutations. The N155H variant was identified in rhesus macaques infected with SHIV 89.6P and treated with L-870812, a napthyridine carboxamide unrelated to raltegravir.23 When introduced into the laboratory strain HIV-1HXB2, a 25-fold reduction in susceptibility to L-870812 was noted. The lack of complete virologic rebound in these animals suggested that the mutant virus was potentially less fit than wild type and/or that the integrase inhibitor was still partly effective against this variant. Interestingly, a persistent antiviral effect was noted in the single patient in this study with the N155H mutation in integrase who remained on treatment despite the presence of resistance-conferring mutations to all 3 components of the treatment regimen. These observations are consistent with continued susceptibility to tenofovir in the presence of K65R + M184V24,25 and the possibility that raltegravir may retain some activity in the presence of the treatment-emergent N155H variant. The N155H mutation confers 15-fold resistance to raltegravir, and the V151I mutation may augment that resistance (data not shown). Of note, changes at codons 163 and 232 in integrase have not been previously described, and their potential contribution to loss of antiviral activity remains obscure.
In general, raltegravir AUC0-12h, Cmax, and C12h were somewhat higher when raltegravir was coadministered with tenofovir and lamivudine. Lamivudine is predominantly renally eliminated by means of active organic cationic secretion and has not been reported to inhibit uridine diphosphate (UDP) glucuronosyltransferases.26 Consequently, the modest increase in raltegravir exposure in part II is likely the result of an interaction between raltegravir and tenofovir. This is consistent with results from a drug interaction study in healthy volunteers demonstrating that raltegravir AUC0-12h and Cmax were modestly higher when coadministered with tenofovir, although the effect on raltegravir C12h was less pronounced.19
All doses of raltegravir, up to 600 mg twice daily, in combination with tenofovir and lamivudine were generally well tolerated. No dose-related toxicities were observed. Drug-related clinical adverse events tended to be less common with raltegravir than with efavirenz. No drug-related serious adverse events were reported, and only 1 patient discontinued raltegravir because of a drug-related adverse event. Neuropsychiatric symptoms are of particular interest in subjects taking efavirenz. These symptoms occur most frequently at the time of initiation of efavirenz dosing and may persist for a period of months or years.27 In the current study, neuropsychiatric symptoms such as abnormal dreams and nightmares were less common in patients treated with raltegravir than with efavirenz.
Laboratory adverse events profiles did not differ substantially between raltegravir and efavirenz, with the exception of serum lipids. Raltegravir was more lipid neutral than efavirenz based on levels of total cholesterol, LDL-C, and triglycerides.
In summary, this report describes the longest duration of treatment (48 weeks) with the integrase inhibitor raltegravir in treatment-naive patients infected with HIV-1. The results of this study demonstrate that raltegravir in combination with tenofovir and lamivudine has rapid, potent, and durable antiretroviral efficacy and a favorable safety profile compared with efavirenz plus tenofovir and lamivudine. Given this profile and these data, the use of raltegravir in combination with nucleoside reverse transcriptase inhibitors (NRTIs) warrants further evaluation as a potential first-line regimen, particularly in patients for whom non-NRTI or protease inhibitor-based therapy may be less than ideal.
METHODS
Study Design
Protocol 004 was a double-blind, randomized, dose-ranging study in treatment-naive HIV-1-infected patients. Part I consisted of 10 days of raltegravir monotherapy in 35 patients.16 Part II examined the safety, tolerability, and efficacy of raltegravir versus efavirenz, in combination with tenofovir and lamivudine, for up to 48 weeks in 30 patients from part I (cohort I) plus 171 patients randomized into part II (cohort II). The study was conducted at 29 sites in the United States, Canada, Latin America, Thailand, and Australia from June 14, 2005, through October 4, 2006, and was approved by the Ethical Review Committee of each participating site; written informed consent was obtained from each patient before study entry.
Patients who entered part II were randomized to raltegravir at a dose of 100, 200, 400, or 600 mg twice daily or to efavirenz at a dose of 600 mg/d, each with tenofovir at a dose of 300 mg/d and lamivudine at a dose of 300 mg/d, through an integrated voice response system with centralized randomization based on a computer-generated allocation schedule. Randomization was stratified by the HIV-1 RNA level at screening (≦50,000 vs. >50,000 copies/mL). Patients who received raltegravir in part I received the same dosage of raltegravir in part II; patients who received placebo in part I received efavirenz in part II. All study personnel remained blinded to treatment allocation; blinding of drug and dosage was accomplished with matching-image placebo tablets of raltegravir and efavirenz.
Physical examinations and blood samples were collected at baseline (day 1); after 2, 4, 8, 12, 16, 24, 32, 40, and 48 weeks of therapy; and 14 days after treatment discontinuation. In cohort I patients, blood samples for pharmacokinetic (PK) evaluations were collected at day 10 in part I and at week 2 in part II as previously described.16
Patients with lack of response (confirmed plasma HIV-1 RNA level >400 copies/mL) or virologic relapse despite compliance with study therapy could be discontinued from study at the discretion of the investigator. Virologic relapse was defined as 2 consecutive measurements (at least 1 week apart) of (1) plasma HIV-1 RNA level >400 copies/mL after an initial response with plasma HIV-1 RNA level <400 copies/mL or (2) >1.0 log10 increase in plasma HIV-1 RNA above the nadir level. In patients displaying virologic failure (HIV-1 RNA level >400 copies/mL at week 24 or early discontinuation, or virologic relapse), genotypic and phenotypic resistance assays (Monogram Biosciences, South San Francisco, CA) were used to evaluate resistance to tenofovir, lamivudine, and efavirenz at the time of failure as compared with baseline. The emergence of resistance to raltegravir was evaluated by genotyping the integrase coding sequence. Patient plasma-derived viral RNA was isolated, and the integrase gene was reverse-transcribed and sequenced using standard methods. Consensus amino acid sequences were determined and compared with pretreatment genotypes.
Study Population
HIV-1-seropositive patients ≥18 years of age were eligible if they had plasma HIV-1 RNA levels ≥5000 copies/mL and CD4+ T-cell counts ≥100 cells/mm3 at screening. Patients were excluded if they had received any prior antiretroviral therapy or adefovir for more than 7 days or if they had documented resistance to tenofovir, lamivudine, and/or efavirenz as determined by viral resistance assay at screening. Additional eligibility criteria were as previously described.16
Study Medication
Raltegravir tablets or matching placebo were taken twice daily (approximately 12 hours apart) without regard to food intake. Tenofovir tablets and lamivudine tablets were taken once daily with the morning dose of raltegravir (or matching placebo). Efavirenz tablets or matching placebo were taken once daily at bedtime on an empty stomach. Patients recorded the dates of study drug doses on diary cards that were returned at each visit and reviewed for completeness and accuracy.
Statistical Analysis
Antiretroviral Activity
Efficacy analyses were based on a modified intent-to-treat (MITT) approach, which included all randomized patients who received at least 1 dose of study medication, regardless of adherence to entry criteria or deviations from the protocol. Cohort II was the primary analysis population, because previous monotherapy may have altered a patient's response to the combination regimen. Cohort I was included in secondary analyses as a sensitivity check of the conclusions. The primary measurement was the proportion of patients achieving a plasma HIV-1 RNA level <400 copies/mL on the AMPLICOR HIV-1 (Roche Molecular Systems, Pleasanton, CA) Monitor Standard assay. Secondary measurements included the proportion of patients achieving a plasma HIV-1 RNA level <50 copies/mL on the UltraSensitive assay (Roche Molecular Systems, Pleasanton, CA) and the change from baseline in CD4+ T-cell count. Weeks 24 and 48 were the primary and secondary time points for efficacy. This was an estimation study only and was not powered for formal efficacy comparisons between raltegravir and efavirenz.
For the proportion of patients achieving HIV-1 RNA levels <400 and <50 copies/mL, the noncompleter = failure (NC=F) approach was used; all missing values attributable to premature discontinuations were considered failures, regardless of the reason for discontinuation and the success/failure status at the time of discontinuation. The difference in proportions between each of the studied doses of raltegravir and control at 24 and 48 weeks was estimated, and the associated 2-sided confidence interval (CI) was derived using the method of Miettinen and Nurminen.20 For change from baseline in CD4+ T-cell count, the observed failure (OF) approach was used; the baseline CD4+ T-cell count was carried forward for patients who discontinued assigned therapy because of lack of efficacy.
Safety
All patients who took study medication were included in the analysis of safety and tolerability, which included all adverse events that occurred while on study therapy or within 14 days after discontinuation. Laboratory values were examined according to the 1992 Division of AIDS (DAIDS) toxicity guidelines for adults (available at: http://rcc.tech-res-intl.com/tox_tables.htm ). The differences in proportions of patients with drug-related adverse events were compared using the 2-tailed Fisher exact test. To establish further whether raltegravir provides a favorable ratio of benefit to risk in treatment-naive patients, post hoc exploration was performed for neuropsychiatric symptoms and changes in lipid profiles.
Role of the Funding Source
This study was designed by the sponsor (Merck and Co., Inc.) in collaboration with external consultants. The data were collected by the clinical site investigators. The sponsor collated the data, monitored the conduct of the study, performed the statistical analyses, and coordinated the writing of the manuscript with all authors. Data were unblinded for statistical analyses after the databases were locked.
|
|
| |
| |
|
|
|