 |
|
|
| |
Preclinical Pharmacokinetic and ADME Characterization of VCH-916, a Novel Non-Nuclesoside HCV NS5B Polymerase Inhibitor
|
| |
| |
Reported by Jules Levin
43rd EASL
April 23-27, 2008
Milan, Italy
N.Chauret , C. Chagnon-Labelle, M. Diallo, J. Laquerre and S. Plante
ViroChem Pharma Inc, 275 Armand-Frappier Blvd, Laval, QC, Canada, H7V 4A7
AUTHOR CONCLUSIONS
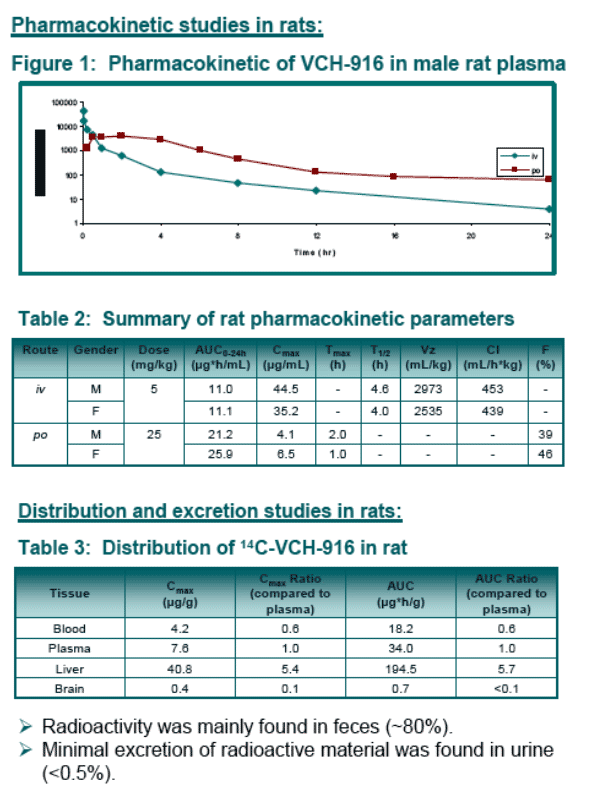
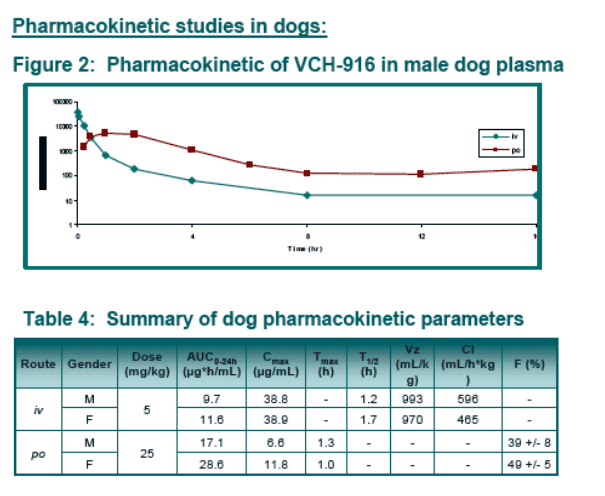
VCH-916 has good oral bioavailability in rat and dog and is mainly eliminated in rat through feces.
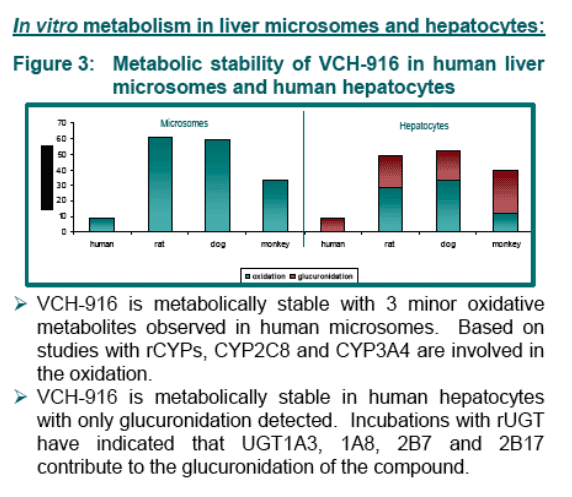
VCH-916 exhibits excellent ADME properties in terms of permeability, distribution in targeted tissue (rat liver) and metabolic pathways.
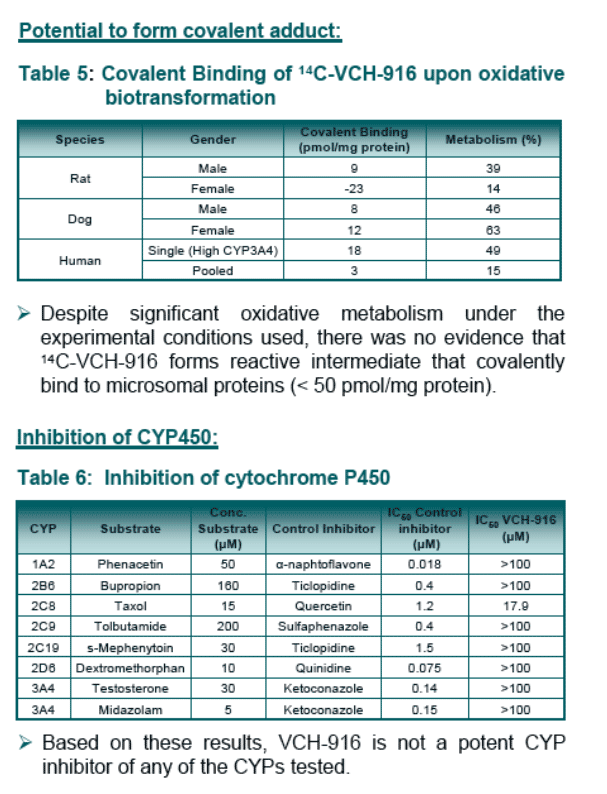
From in vitro studies, several pathways (oxidation and glucuronidation) with several enzymes (CYP and UGT) are involved in the biotransformation of VCH-916. There is no evidence for the formation of potential reactive intermediates upon bio-activation.
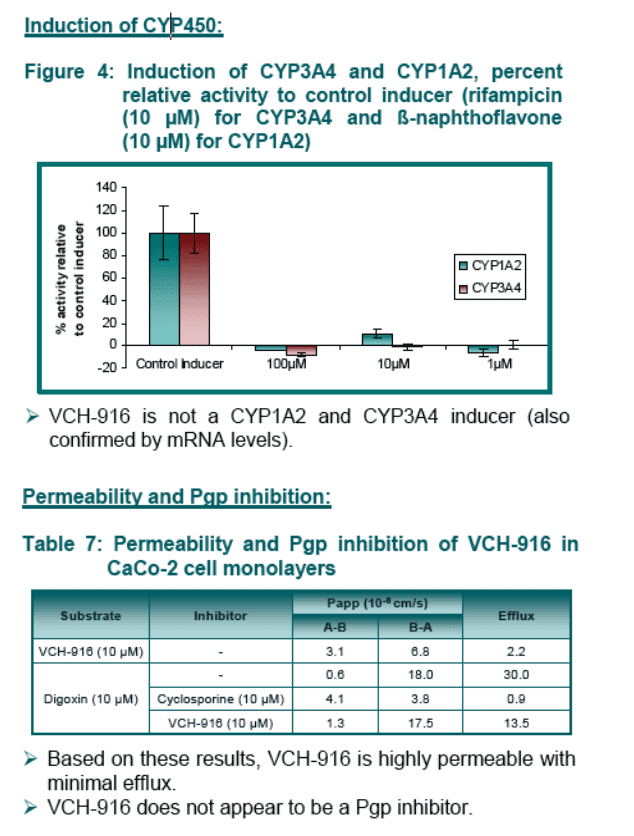
Based on in vitro studies, it is unlikely that VCH-916 will cause drug interactions in humans via CYP inhibition, CYP induction or Pgp inhibition.
ABSTRACT
Background and aims: VCH-916 is a novel nonnucleoside HCV RNA polymerase inhibitor. It has demonstrated sub-micromal IC50s against the HCV replicons of genotype 1a and 1b. It is currently being evaluated for safety/tolerability, pharmacokinetics and antiviral efficacy in chronically infected HCV patient. The objective of these studies was to characterize the pharmacokinetic profile in relevant animal models and ADME properties of VCH-916.
Methods: Pharmacokinetic studies were conducted following iv and po administration of VCH-916 in rats and dogs. The routes of excretion and distribution of the compound in the liver were determined in rats following oral administration of 14C-VCH-916. The absorption, metabolic stability, covalent binding, protein binding and the potential of VCH-916 to cause Pgp inhibition, CYP450 inhibition/induction were evaluated in vitro in systems derived from human and/or animal species.
Results: VCH-916 was relatively stable in human microsomes and hepatocytes. Both oxidation and glucuronidation pathways were observed. Phenotyping studies indicated that several enzymes were involved in the biotransformations of VCH-916 (CYP2C8, 3A4, UGT1A3,1A8, 2B7, 2B17). No covalent binding to microsomal proteins upon oxidative bio-activation was noticed. In all species, plasma protein binding was extensive (>98%). VCH-916 demonstrated limited potential to cause human CYP inhibition or induction. The absorption potential of VCH-916 was shown to be high based on its high permeability in CaCo-2 cell and lack of recognition by intestinal efflux proteins. In addition, VCH-916 had no significant effect of digoxin permeability. In rat and dog, VCH-916 displays low total body clearance with excellent oral bioavailability (>40%). After oral administration of 14C-VCH-916 to rats, recovery of radioactivity was almost complete (80%) 24-hr post-dosing with the majority of the dose excreted in feces. The exposure of 14C-VCH-916 in liver was 5-fold higher than in plasma.
Conclusion: VCH-916 is orally bioavailable and exhibits excellent ADME properties in terms of permeability and metabolic profile. VCH-916 is neither a CYP inhibitor/inducer or Pgp inhibitor reducing the likehood of VCH-916 to be involved in drug interactions. From these data, a favorable clinical pharmacokinetic profile is expected in humans.
OBJECTIVE
Evaluate the pharmacokinetic profiles of VCH-916 in rat and dog.
Determine the excretion and distribution of 14C-VCH-916 in rats.
Determine the in vitro metabolic characteristics of VCH-916 in animals and in humans.
Evaluate, via in vitro studies, the potential of VCH-916 to cause drug-drug interactions through CYP and Pgp enzymes.
EXPERIMENTAL
Plasma protein binding: The extent of binding of 14C-VCH-916 to plasma protein was determined by ultrafiltration using M.W. cutoff membranes of 30,000.
Pharmacokinetic profiles: VCH-916 was administered to rats and dogs orally (in 0.5% CMC) and intravenously (in 30% PEG) at 25 mg/kg and 5 mg/kg respectively.
Excretion and liver distribution: 14C-VCH-916 was administered orally to rats at 25 mg/kg (330 _Ci/kg) and selected tissues or fluids were collected over a 24-hr period.
In vitro metabolism studies: VCH-916 (20 _M) was incubated in liver microsomes (1.0 mg/mL) obtained from rat, dog, monkey and human for 1.5 hours in presence of NADPH (1.5 mM). VCH-916 (10 _M) was also incubated in human, rat, dog and monkey hepatocytes (106 cells/mL) for 2 hours. Incubations in microsomes expressing individual CYP (20 pmol) or UGT (0.5 mg/mL) using NADPH (1.5 mM) or UDPGA (1.5 mM) as co-factor, respectively, were also conducted. The microsomal and hepatocyte incubates were analysed by HPLC/UV and MS for parent disappearance and for metabolite formation. In addition, microsomal incubation (2 mg/mL protein, 1.5 mM NADPH) were performed with 14C-VCH-916 (19.2 uM, 0.5 uCi) to determine if VCH-916 could form covalent adduct with microsomal protein. The radioactivity in the protein pellet was analysed by total count analysis.
In vitro CYP inhibition: The effect of VCH-916 on CYP450 activity was studies in human liver microsomes supplemented with NADPH (1 mM) using selective substrates.
In vitro CYP induction: The effect of VCH-916 on the CYP3A4 and 1A2 activity and mRNA levels was evaluated in cultured human hepatocytes (140,000 cells, 48-hr).
Permeability and in vitro inhibition of Pgp: The permeability of VCH-916 and the effect of VCH-916 on the efflux of digoxin was studied in CaCo-2 cells monolayers grown to confluence on polycarbonate membranes.
Results and Discussion

|
| |
|
|
|
|
|