| |
Hemochromatosis Gene Polymorphisms, Mitochondrial Haplogroups, and Peripheral Lipoatrophy during ART
|
| |
| |
The Journal of Infectious Diseases March 3, 2008
(See the editorial commentary by Lichtenstein on pages XXX-XXX.)
Todd Hulgan,1 Pablo Tebas,2 Jeffrey A. Canter,1 Kathleen Mulligan,3,a David W. Haas,1 Michael Dube,4 Steven Grinspoon,5 Gregory K. Robbins,5 Alison A. Motsinger,1 and Asha R. Kallianpur,1 for the
AIDS Clinical Trials Group 384 and A5005s Study Teams
1Vanderbilt University School of Medicine, Nashville, Tennessee; 2University of Pennsylvania, Philadelphia, Pennsylvania; 3University of California at San Francisco, San Francisco, California; 4Indiana University School of Medicine, Indianapolis, Indiana; and 5Massachusetts General Hospital, Harvard University, Boston, Massachusetts
"The present study suggests that the prevalent HFE 187C>G polymorphism is associated with reduced risk of lipoatrophy (determined by DEXA). Further studies are needed to replicate this association in other cohorts and to determine the mechanism(s) underlying this association. The trend toward protection from fat loss associated with European haplogroup J also suggests the potential importance of mitochondrial genetic variation. Common genetic variations in iron-regulatory and mitochondrial genes may contribute to a better understanding of the pathogenesis of lipoatrophy, potentially fostering new prevention and treatment strategies."
ABSTRACT
Background. Antiretroviral therapy (ART)-associated lipoatrophy involves mitochondrial dysfunction. Iron metabolism impacts mitochondrial function and oxidative stress. Mitochondrial haplogroups and hemochromatosis gene (HFE) polymorphisms have been associated with ART-induced neuropathy.
Methods. The AIDS Clinical Trials Group 384 study randomized ART-naive individuals to receive didanosine-stavudine or zidovudine-lamivudine, combined with efavirenz and/or nelfinavir. Substudy A5005s evaluated fat distribution by dual-energy X-ray absorptiometry (DEXA). We characterized HFE polymorphisms 845G>A and 187C>G and European mitochondrial haplogroups in A5005s participants who consented to genetic analyses.
Results. Among 96 participants (58% were white, and 10% were female) with baseline and 48 or 64 week DEXA data, the median limb fat change was -8.8% (interquartile range, -28.7% to +15.6%). HFE 187C/G heterozygotes (n=23) had less limb fat loss than 187C/C homozygotes (n=71) (+6.1% vs. -12.5%; p=.02 ) and were less likely to develop lipoatrophy after adjustment for age, sex, race, and ART randomization (odds ratio, 0.31; 95% confidence interval, 0.10-0.95; ). Among non-Hispanic white participants, median limb fat change was +26.1% among 5 participants with mitochondrial haplogroup J, compared with -9.7% among 49 participants with other mitochondrial haplogroups (p=.07).
Conclusions. HFE 187C>G and, possibly, mitochondrial haplogroup J gave relative protection against lipoatrophy during ART in A5005s. These associations should be replicated in other studies.
Potent antiretroviral therapy (ART) has reduced morbidity and mortality due to acquired immunodeficiency syndrome (AIDS). Preferred initial therapies for human immunodeficiency virus type 1 (HIV) infection include 2 nucleoside reverse-transcriptase inhibitors (NRTIs) plus either an HIV protease inhibitor or a nonnucleoside reverse-transcriptase inhibitor [1]. Unfortunately, NRTI inhibition of mitochondrial DNA (mtDNA) polymerase-ϒ may contribute to treatment-limiting toxicities [2].
Loss of subcutaneous fat from the extremities and face (i.e., lipoatrophy) is believed to result from mitochondrial dysfunction due in part to chronic NRTI exposure [3]. The thymidine-analogue NRTIs (stavudine or zidovudine) are most frequently associated with lipoatrophy, and switching from stavudine or zidovudine to NRTIs with lower affinity for mtDNA polymerase can slow progression of lipoatrophy [4-6]. Other putative risk factors for lipoatrophy include male sex, white race, lower pre-ART weight, greater cumulative NRTI exposure, more-advanced HIV disease, and a tumor necrosis factor (TNF)-α gene promoter polymorphism [7-9]. Recent data have also suggested use of the protease inhibitor nelfinavir as a possible risk factor [10].
Lipoatrophy may involve recruitment of activated monocyte/macrophages to adipose tissues, possibly in response to disease-associated and/or drug-associated mitochondrial damage [11]. Oxidative stress induced by mitochondrial dysfunction and macrophage-mediated cytokine/chemokine release may promote these toxic complications. In lipoatrophy the exuberant inflammatory response is associated with adipocyte toxicity and apoptosis [12]. Only some persons exposed to NRTIs develop significant lipoatrophy, suggesting a genetic predisposition among certain individuals. Studies to identify genetic susceptibility factors in lipoatrophy may improve drug selection at the time of ART initiation and lead to new strategies for preventing and managing this complication.
Among AIDS Clinical Trials Group (ACTG) 384 study participants who received ART that included didanosine and stavudine, we previously observed relative protection from peripheral neuropathy in persons heterozygous for the hemochromatosis (HFE) mutation 845G>A and a trend toward protection from this complication in carriers of another common variant, HFE 187C>G [13]. Hereditary hemochromatosis is a genetic iron-overload disorder, usually resulting from a homozygous missense mutation (nucleotide 845G>A) that substitutes tyrosine for cysteine at position 282 in the HFE gene [14]. Although 845G>A heterozygotes rarely develop hemochromatosis, they exhibit increased iron stores and altered macrophage iron transport [15, 16]. The more prevalent HFE 187C>G polymorphism, which substitutes aspartate for histidine at position 63, is also associated with increased iron indices but is rarely the sole cause of iron overload [14, 17, 18]. The reduced toxic neuropathy observed in carriers of HFE polymorphisms may occur by a number of mechanisms, which we have outlined previously [13]. The macrophage iron transport defect associated with the 845G>A polymorphism is known to lead to a lower-than-normal iron content in these cells; macrophage iron deficiency may, in turn, result in less oxidative nerve injury and neuronal apoptosis during macrophage-mediated inflammation. Alternatively, an improved supply of iron to metabolically active cells such as neurons may benefit mitochondrial function in HFE carriers, rendering these individuals less susceptible to NRTI-associated mitochondrial toxicities.
The human mitochondrial genome comprises circular, double-stranded DNA (i.e., mtDNA) that encodes ribosomal RNAs, transfer RNAs, and 13 polypeptides that are essential for oxidative phosphorylation. Stable single-nucleotide polymorphisms (SNPs) in the mitochondrial genome define mitochondrial haplogroups, the distribution of which has been used to map prehistoric human migrations [19, 20]. We previously identified an association between European mitochondrial haplogroup T and peripheral neuropathy among participants in ACTG 384 [21].
We hypothesized that HFE polymorphisms and/or stable SNPs in the mtDNA, such as those defining mitochondrial haplogroups, modulate susceptibility to lipoatrophy. This study, involving participants from the A5005s metabolic substudy of ACTG 384, examined relationships between HFE polymorphisms, mitochondrial haplogroups, and limb fat changes during NRTI-containing ART.
Discussion
Among participants in the A5005s metabolic substudy of ACTG 384, heterozygous carriers of the HFE 187C>G polymorphism receiving ART that included didanosine-stavudine or zidovudine-lamivudine experienced significantly less limb fat loss than noncarriers and in fact had a median gain in limb fat of >6%. The association was independent of age, race, sex, and study drug randomization groups. This is the first study to demonstrate that a genetic polymorphism affecting iron transport may modulate susceptibility to lipoatrophy. Among white study participants, mitochondrial haplogroup J also tended to be associated with reduced limb fat loss, compared with non-J haplogroups. These findings suggest potential links between lipoatrophy and variations in iron transport, the inflammatory response, and mitochondrial genes.
In the United States, the prevalence of HFE 845G>A heterozygosity is 10%-12% among white persons and 2%-3% among African Americans; HFE 187C>G heterozygosity is seen in 23%-24% and 5%-6% of these populations, respectively [26]. Although these common HFE gene variants have been shown to impact iron transport, their individual effects may differ qualitatively and by cell type [14, 27-29]. The hemochromatosis protein (Hfe) is an atypical class I-like major histocompatibility complex molecule that is expressed on intestinal crypt cells and on cells of monocyte/macrophage lineage. Hfe is involved in the regulation of iron absorption and iron metabolism, but its mechanisms of action and direct effects remain unclear [14, 30-32]. A major role of Hfe that has emerged recently is regulation of hepcidin synthesis. Hepcidin is an iron-regulatory peptide normally released by the liver in response to iron loading and inflammatory stimuli such as IL-6 and TNF-α [33]. Hepcidin also inhibits gut iron absorption and macrophage iron efflux, leading to iron accumulation within monocytes/macrophages, low serum iron concentrations, and the anemia of chronic inflammation, presumably as an antimicrobial defense mechanism [27]. HFE position 845A/A homozygosity has been linked to inappropriately low hepcidin levels and iron-poor tissue macrophages despite iron overload [32, 34]. Whether hepcidin expression and macrophage iron transport are similarly dysregulated in HFE position 187C/G heterozygotes is unclear.
Monocytes and macrophages, via their iron recycling role, are critically important in iron homeostasis, and they may also mediate inflammatory damage to target tissues in lipoatrophy and peripheral neuropathy [11, 35]. Macrophages accumulate iron during HIV-1 infection and other chronic inflammatory and infectious diseases, consistent with the action of proinflammatory cytokines and hepcidin. These observations are of particular interest in light of recent evidence that the HIV-1 Nef protein specifically down-regulates Hfe expression on macrophages derived from healthy individuals, altering macrophage iron homeostasis and possibly benefiting viral replication in these cells [36]. HIV-infected macrophages derived from HFE 845A/A homozygotes, which express variant Hfe, do not show these effects. In a mouse model, the degree of surface Hfe expression influenced macrophage iron transport as well as hepatic hepcidin synthesis, consistent with an indirect role for HFE in inflammation [34].
Several potential mechanisms might therefore explain the relative protection against lipoatrophy associated with HFE 187C>G in A5005s. If monocytes/macrophages are recruited to adipose tissue in response to mitochondrial damage and local oxidative injury, the ensuing release of proinflammatory cytokines may promote adipocyte toxicity [12, 37]. Chronic exposure to proinflammatory cytokines and chemokines, many of which are produced by adipocytes themselves (adipokines) and have autocrine or paracrine effects (e.g., IL-6 and TNF-α), can induce adipocyte apoptosis and lipolysis in vitro [38-40]. Adipocyte-mediated TNF-α production has also been shown to upregulate macrophage-chemoattractant protein type 1 [37, 38]. Hepcidin levels may also be elevated in HIV-1-infected individuals because of the inflammatory milieu. A vicious cycle of oxidative stress due to increased macrophage iron content, release of inflammatory mediators, and further recruitment of monocytes/macrophages to adipose tissue may lead to lipoatrophy. Conversely, if HFE 187C>G, like 845G>A, is associated with reduced hepcidin expression and diminished monocyte/macrophage iron content, these changes may lead to reduced inflammation, oxidative stress, and adipocyte death. There have also been conflicting reports regarding TNF-α levels in HFE carriers in response to inflammatory stimuli [41, 42]; lower levels of TNF-α would limit macrophage recruitment to fat and, presumably, lower hepcidin levels. Finally, increased circulating iron in carriers of HFE variants caused by increased gut iron absorption and macrophage iron efflux may be important in preventing or limiting lipoatrophy. An adequate cellular iron supply may be critical for preadipocyte differentiation [43] and may thus be needed to compensate for adipocyte loss due to mitochondrial damage. Of note, rats fed iron-deficient diets undergo increased lipolysis and adipocyte apoptosis, with the threshold for apoptosis varying between fat depots [44].
Mitochondrial dysfunction underlies many adverse effects commonly attributed to NRTIs, with mtDNA depletion due to inhibition of host mtDNA polymerase-ϒ proposed as an underlying mechanism [45]. There is thus ample rationale for a link between mitochondrial genetics and NRTI toxicity. However, attempts to correlate NRTI cellular toxicity with changes in mtDNA content have yielded variable results, suggesting that other factors likely contribute to the phenotypic expression of these toxicities. Epidemiological evidence for functional differences between mitochondrial haplogroups has emerged from studies of neurodegenerative disorders. European haplogroup J (characterized by polymorphisms at positions 10398A>G and 13708G>A) has been associated with longevity [46], relative protection against Parkinson disease [47], and expression of Leber's hereditary optic neuropathy, a cause of adult-onset blindness [48]. Molecular mechanisms linking mtDNA polymorphisms with these phenotypes and with lipoatrophy are unknown but may reflect impaired energy production. The effects of mtDNA polymorphisms on oxidative phosphorylation efficiency may be unmasked by exposure to stressors such as NRTIs and/or HIV infection, and variants such as those defining haplogroup J could explain the expression of clinical phenotypes via DNA polymerase-ϒ-dependent and -independent mechanisms. The greatest diversity of mtDNA is found in Africa, where >150 polymorphisms define >100 haplogroups [49]. European haplogroups are much less diverse; hence, haplogroup assignment was limited to white participants in this pilot study.
Some differences should be noted between our results and those of previous studies. In adjusted analyses, we did not find a statistically significant association between lipoatrophy and randomization to the didanosine-stavudine arm or with nelfinavir use, whereas both factors conferred excess risk in the primary analysis of A5005s [10]. This discrepancy may reflect the much smaller sample size in the present study, which was limited to A5005s participants who provided consent for genetic analyses. However, it is also possible that including HFE genotype in the multivariable model weakened the association between specific drugs and lipoatrophy. Supporting this explanation is the observation that relative limb fat loss tended to be greater in HFE 187C/C homozygous individuals randomized to receive zidovudine-lamivudine than in HFE 187C/G heterozygous individuals randomized to receive didanosine-stavudine, a combination that is no longer recommended for use because of high rates of mitochondrial toxicity [1].
There were few 845A allele carriers in our study population. Although differences based on the major 845G>A polymorphism were not statistically significant in univariate or adjusted analyses, these 6 individuals did not appear to receive any protection from limb fat loss. Finally, since the HFE and HLA-A loci are in linkage disequilibrium, we cannot exclude the possibility that HLA-A haplotypes or other genes within the major histocompatibility complex explain this association. Indeed, a TNF-α promoter polymorphism has been associated with lipodystrophy in HIV-infected individuals [8, 9], and TNF gene polymorphisms may modify the iron overload phenotype in HFE position 845A/A homozygotes, although published studies are inconsistent on this point [41, 42, 50].
The present study suggests that the prevalent HFE 187C>G polymorphism is associated with reduced risk of lipoatrophy (determined by DEXA). Further studies are needed to replicate this association in other cohorts and to determine the mechanism(s) underlying this association. The trend toward protection from fat loss associated with European haplogroup J also suggests the potential importance of mitochondrial genetic variation. Common genetic variations in iron-regulatory and mitochondrial genes may contribute to a better understanding of the pathogenesis of lipoatrophy, potentially fostering new prevention and treatment strategies.
Patients and Methods
Study participants and design.
ACTG 384 was a randomized, multicenter clinical trial to evaluate initial HIV treatment strategies in adults [22, 23]. Briefly, volunteers were enrolled in the United States and Italy in 1998 and 1999. Eligibility criteria included a plasma HIV-1 RNA load of 500 copies/mL and <7 days of prior ART. Patients were randomized to receive 3- or 4-drug therapy with didanosine-stavudine or zidovudine-lamivudine in combination with efavirenz, nelfinavir, or both. Regimen failure was defined by virologic and toxicity-related criteria, and the primary end point was time to failure of the second regimen or discontinuation of all study medications. Self-reported race/ethnicity categories were "white, non-Hispanic," "black, non-Hispanic," and "Hispanic"; we hereafter refer to these groups as white, black, and Hispanic, respectively.
The ACTG A5005s substudy (described elsewhere [10]) enrolled 334 participants from ACTG 384 at 23 clinical trial sites in the United States. Persons with diabetes were excluded. A total of 157 subjects from 18 sites underwent whole-body, dual-energy X-ray absorptiometry (DEXA) that was analyzed centrally at baseline and every 16 weeks thereafter. Ninety-six individuals had specimens available from the ACTG Human DNA Repository (protocol A5128 [24]) at the Vanderbilt Center for Human Genetics Research (Nashville, TN) and had DEXA results available at baseline and at week 48 or 64 after the initiation of ART. Many ACTG 384/A5005s study participants did not participate in A5128 because A5128 did not open for accrual at participating institutions until almost 4 years after ACTG 384 had begun. Demographic and clinical characteristics of participants from ACTG 384 who contributed DNA for genetic analyses were similar to the characteristics of those who did not [21].
The 384, A5005s, and A5128 studies were approved by the institutional review boards at each site, and all participants provided written informed consent. The Vanderbilt Committee for the Protection of Human Subjects and the ACTG approved the use of DNA.
Study treatments.
Participants received the following open-labeled NRTIs: didanosine (400 mg or 250 mg daily on the basis of weight; enteric-coated tablets were available on request during the final year of the study) and stavudine (40 mg or 30 mg twice daily on the basis of weight); or zidovudine (300 mg) and lamivudine (150 mg), administered as a fixed-dose combination twice daily. In addition, participants received efavirenz (600 mg once daily) and/or nelfinavir (1250 mg twice daily) in a double-blinded fashion with matching placebos. Two NRTI substitutions (stavudine for zidovudine, and lamivudine for didanosine) were allowed during the study because of intolerance.
Human genotyping
Mitochondrial and HFE genotyping was performed as previously described [13, 21]. Genomic DNA was extracted from whole blood, using Puregene (Gentra Systems). Genotyping was accomplished with the ABI Prism 7900 HT Sequence Detection System (Applied Biosystems). Customized TaqMan assays were used to genotype both HFE loci 845G>A (rs1800562) and 187C>G (rs1799945) and SNPs at mtDNA positions 1719G>A, 4580G>A, 7028C>T, 8251G>A, 9055G>A, 10398A>G, 12308A>G, 13368G>A, 13708G>A, and 16391G>A. Proprietary probe and primer sequences, cycling, and scanning conditions are provided in table 1. European mitochondrial haplogroups were assigned using previously described methods [19]. Data from patient samples were analyzed using ABI Sequence Detection System software, version 2.1. Genotypes were confirmed by visual inspection of plots. Laboratory personnel who had no knowledge of clinical phenotypes performed the genotyping.
Results
A total of 204 participants from A5005s were included in this genetic analysis, of whom 105 had at least 1 DEXA result. Our primary analysis focused on 96 individuals with HFE genotype data as well as baseline and either 48-week (10 participants) or 64-week (86 participants) DEXA results. Of these individuals, 58% were white, 10% were female, the median age was 36 years, and the median body mass index (defined as the weight in kilograms divided by the square of the height in meters) at baseline was 23.7 (table 2). The median baseline CD4 lymphocyte count was 242 cells/mm3 (interquartile range [IQR], 81-415 cells/mm3), and the plasma HIV-1 RNA concentration was 5.1 log10 copies/mL (IQR, 4.6-5.7 log10 copies/mL). A total of 56% had been randomized to receive didanosine-stavudine in their initial regimen. There were no significant differences at baseline between study subjects with DEXA and HFE genotyping data available and all A5005s subjects who participated in A5128 (table 2).
Overall, participants lost limb fat during the study (table 2), with a median change at 48 or 64 weeks of -0.4 kg (IQR, -1.8 to +0.9 kg) or a median relative change of -8.8% (IQR, -28.7% to +15.6%).
Eleven participants (11%) were heterozygous for HFE 845G>A, and 23 (24%) were heterozygous for HFE 187C>G. None were homozygous at either allele. The frequencies of HFE 845A and 187G alleles in this study population did not deviate from expected race-specific values published previously [25, 26]. Position 845G/A heterozygotes did not differ from 845G/G homozygotes with respect to change in limb fat (-13.4% vs. -8.2%; p=.71) or lipoatrophy (unadjusted OR, 1.5; 95% CI, 0.42-5.24; p=.54). However, position 187C/G heterozygotes had significantly less limb fat loss at 48 or 64 weeks, compared with 187C/C homozygotes (+6.1% vs. -12.5%; p=.02) (table 3) and were less likely to develop lipoatrophy (10% limb fat loss from baseline; unadjusted OR, 0.32; 95% CI, 0.11-0.92; p=.03). In unadjusted analyses, randomization to a nelfinavir-containing regimen was the only other variable associated with increased lipoatrophy (OR, 2.63; 95% CI, 1.05-6.60; p=.04). Randomization to didanosine-stavudine was also associated with development of lipoatrophy, but the association did not reach statistical significance (OR, 2.09; 95% CI, 0.91-4.78; p=.08).
Table 3. Limb fat changes and lipoatrophy over 48 or 64 weeks, by hemochromatosis gene (HFE) genotype and mitochondrial haplogroup.
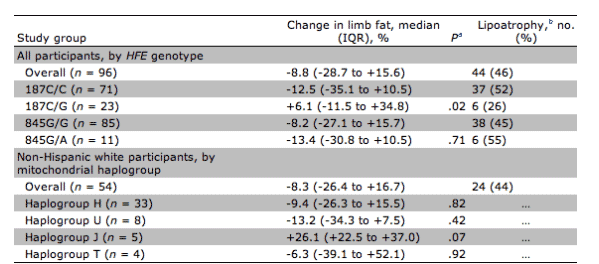
aData were calculated by the Wilcoxon rank sum test and involve comparisons between 187C/G and 187C/C, 845G/A and 845G/G, and individual haplogroups versus all other haplogroups.
bDefined as a >10% decrease in limb fat.
Among non-Hispanic whites, 54 (96%) were successfully genotyped for European mitochondrial haplogroup classification (table 3). The 5 haplogroup J individuals had a median limb fat change of +26% (IQR, +23% to +37%), compared with -9% (IQR, -26% to +4%) among non-J individuals (p=.07). Other haplogroups were H (33 participants), T (4 participants), U (8 participants), and other non-J haplogroups (4 participants); none of these were associated with a statistically significant change in limb fat.
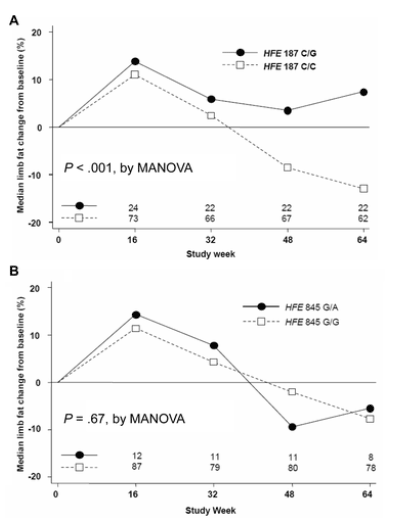
As presented in table 4, HFE 187C>G remained associated with a reduced risk of lipoatrophy after adjustment for age, sex, race/ethnicity, and randomized study drugs (adjusted OR, 0.31; 95% CI, 0.10-0.95; p=.04). There was a trend toward association with nelfinavir in the adjusted model. We also used a random-effects MANOVA model to assess differences in limb fat changes over time. The overall pattern of limb fat change among all subjects with available DEXA results, adjusted for baseline demographic characteristics, race/ethnicity, and study drug randomization, differed significantly between HFE position 187C/G heterozygotes and C/C homozygotes (p<.001, by MANOVA; (figure 1A) but not by 845G>A genotype (, by MANOVA; figure 1B). Similar results were obtained by MANOVA stratified by race/ethnicity and NRTI study drugs at randomization (data not shown).
Table 4. Univariate and multivariate logistic regression analyses of factors associated with lipoatrophy.
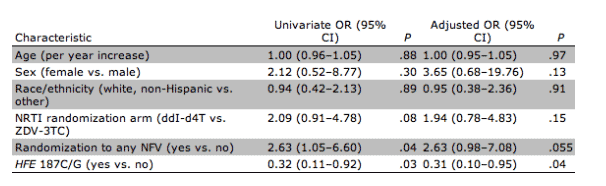
Figure 1. Overall pattern of limb fat changes by hemochromatosis gene (HFE) genotype 187C>G (A) and HFE 845G>A (B). Values are based on dual-energy X-ray absorptiometry findings. P values were adjusted for age, race/ethnicity, sex, nucleoside reverse-transcriptase inhibitor randomization arm, and randomization to any nelfinavir versus no nelfinavir. MANOVA, multivariable analysis of variance.
When study participants were analyzed by NRTI randomization group and HFE genotype, median changes in limb fat at 48 or 64 weeks were greatest among individuals randomized to receive didanosine-stavudine who were position 187C/C homozygotes (median change, -15.0% [IQR, -38% to +3%]). Limb fat loss was of intermediate severity among position 187C/C homozygotes who received zidovudine-lamivudine (median change, -10%) and among position 187C/G heterozygotes who received didanosine-stavudine (median change, -2%). Position 187C/G heterozygotes who were randomized to receive zidovudine-lamivudine exhibited a net gain in limb fat (median change, +10%). Subgroup comparisons by randomized treatments and HFE genotype did not attain statistical significance (p=.15 for didanosine-stavudine and p=.06 for zidovudine-lamivudine).
|
|
| |
| |
|
|
|