| |
ART Effect on Immune Activation Reduces Cerebrospinal Fluid HIV-1 Infection: even treatment failures has positive impact on the brain
|
| |
| |
JAIDS Journal of Acquired Immune Deficiency Syndromes:Volume 47(5)15 April 2008pp 544-552
Sinclair, Elizabeth PhD*; Ronquillo, Rollie BS*; Lollo, Nicole BA; Deeks, Steven G MD*; Hunt, Peter MD*; Yiannoutsos, Constantin T PhD; Spudich, Serena MD; Price, Richard W MD
From the *Department of Medicine, University of California at San Francisco, San Francisco, CA; Department of Neurology, University of California, San Francisco, CA; and the Division of Biostatistics, Department of Medicine, Indiana University School of Medicine, Indianapolis, IN.
This study sought: "To explore whether T-cell activation relates to the magnitude of CSF HIV-1 infection and whether reduced activation associates with treatment effects in this compartment".
"......Cross-sectional analysis of 14 HIV-negative subjects and 123 neuroasymptomatic HIV-1-infected subjects.....
......CNS HIV-1 infection is an important component of systemic infection and a critical target of therapy. These studies suggest that the level of systemic immune activation is an important modulator of this infection and that its downregulation by ART may contribute to controlling HIV-1 in this compartment and explain the better-than-predicted responses of CSF HIV-1 to ART and both the preventative40 and therapeutic41 effects of treatment on ADC, although importantly the current study was confined to subjects without neurological impairment.......
.... Compared to the offs (median 47.1%) (Fig. 2A), blood CD8 T-cell activation was clearly reduced in the failures, >500 c/ml on HAART, (median 34.2%), with the successes (<500 c/ml) showing further reduction (median 21.5%), yet not equal to the HIV-negative controls.... Although CD8 T-cell activation was somewhat higher in the CSF than in blood, this same comparative order was noted (Fig. 2B): medians for offs 61.3%, failures 39.1%, successes 26.7%, and HIV-negative controls 23.9%...... In both compartments there was a downward shift in CD8 T-cell activation in the face of a given level of resistant HIV-1 compared to wild-type virus......
......systemic HIV-1 replication is the prime mover in this interaction..... this model suggests that reducing systemic immune activation with ART may contribute to suppressing CSF infection, not only in the failures but also in the successes. (from Jules: these study results suggest to me that perhaps the lower the blood viral load the better, undetectable is better in terms of controlling CSF HIV levels and in reducing immune activiation; thus, HAART interruptions may increase HIV levels in the brain).....
......The resultant final model was highly significant (P < 0.0001; r2 = 0.814) and included as significant predictors of CSF HIV RNA levels the variables plasma HIV-1 RNA (P < 0.001), CSF neopterin (P < 0.001), and blood CD8 activation (P = 0.015).....
......Although the plasma HIV-1 RNA levels were not significantly different between the offs and failures (median 0.35 log10 copies/mL higher in offs than failures; Table 1), the CSF HIV-1 RNA levels were significantly higher in the offs than the failures (difference in medians of 1.65 log10, P < 0.001)......
.....The levels of activation of CSF CD8 cells and, to a lesser extent, CD4 cells were highly correlated with their blood counterparts across the subject groups and the range of cell activation.....
.....HIV-1 infection and treatment outcome had a marked effect on T-cell activation in both blood and CSF, with a stepwise decrease in the level of blood CD8 CD38/HLA-DR coexpression with partial and full viral suppression. Thus, although suppressive therapy had a greater impact, even failed treatment related to the development of drug resistance had a substantial and statistically significant effect, confirming previous reports related to blood cells19,28 and now extending this finding to CSF T cells..... wild-type virus is associated with a higher level of activation than resistant virus at a given level of plasma viremia......"
STUDY BACKGROUND
HIV-1 reaches the central nervous system (CNS) early during primary infection1-3 and can be detected in the cerebrospinal fluid (CSF) throughout the course of chronic untreated systemic infection in the great majority of those infected.4,5 Although asymptomatic and seemingly benign in most individuals, CNS infection evolves in some to a more invasive encephalitis, manifesting clinically as AIDS dementia complex (ADC).6-9 Chronic HIV-1 infection within the isolated body compartment of the CNS (including leptomeninges, brain perivascular spaces, and brain parenchyma) may also have broader implications for viral persistence and responses to antiretroviral therapy (ART). Because the blood-brain barrier (BBB) and blood-CSF barriers (BCB) restrict antiviral drug penetration, it is feared that compartmentalized infection in the CNS can evade therapeutic drug concentrations, allow viral escape, and promote evolution of drug-resistant mutants.10-12
We recently reported a cross-sectional study showing that not only was CSF HIV-1 usually controlled in individuals with suppressed plasma HIV-1, but that treated subjects with continued viremia due to drug resistance benefited virologically in the CSF more than plasma.13 Thus, though subjects failing treatment had plasma HIV-1 levels similar to those off treatment, their CSF viral HIV-1 RNA concentrations were 10-fold lower. We referred to this enhanced treatment-mediated benefit in drug-resistant subjects as a disproportionate therapeutic effect on CSF. The overall favorable responses in CSF were consistent with other reports, including longitudinal studies of treatment initiation.5,14-16 The disproportionate effect on CSF HIV-1 RNA in resistant subjects was also consonant with observations in earlier longitudinal studies of treatment in resistant subjects who showed a continued fall in CSF HIV-1 despite a plateau or a rebound in plasma HIV-1 RNA5 and with greater CSF rebound than plasma HIV-1 rebound in subjects undergoing treatment interruption in the setting of virologic failure.17
Among the possible mechanisms mediating this disproportionate effect on CSF infection, we hypothesized that reduced immune activation accompanying drug-resistant viremia might play a role.13,18,19 CD8, and to a lesser extent CD4, T-cell activation is a strong independent predictor of CD4 T-cell loss and overall prognosis in HIV-1 infection,20-22 and attenuation of immune activation may be an important contributor to treatment responses. In the setting of drug resistance, reduced T-cell activation can predict slower CD4 T-cell decline despite continued plasma viremia.22 To explore whether T-cell activation relates to the magnitude of CSF HIV-1 infection and whether reduced activation associates with treatment effects in this compartment, we used flow cytometry to examine surface coexpression of CD38 and HLA-DR as indices of T-cell activation in CSF and blood in relation to HIV-1 CSF infection in our subject cohort. We emphasized the effect of antiretroviral therapy on the relation between CD8 activation to CSF HIV-1 RNA levels, but also measured CD4 activation. These studies were undertaken in the broader context of a program to characterize CSF T cells and how they relate to their blood counterparts.23,24
Abstract
Objective: To define the effect of antiretroviral therapy (ART) on activation of T cells in cerebrospinal fluid (CSF) and blood, and interactions of this activation with CSF HIV-1 RNA concentrations.
Design: Cross-sectional analysis of 14 HIV-negative subjects and 123 neuroasymptomatic HIV-1-infected subjects divided into 3 groups: not on ART (termed offs), on ART with plasma HIV-1 RNA >500 copies/mL (failures), and on ART with plasma HIV-1 RNA ��500 copies/mL (successes). T-cell activation was measured by coexpression of CD38 and human leukocyte antigen DR (HLA-DR). Other measurements included CSF neopterin and white blood cell (WBC) counts.
Results:
CD8 T-cell activation in CSF and blood was highly correlated across all subjects and was highest in the offs, lower in the failures, and lower still in the successes.
While CD8 activation was reduced in failures compared to offs across the range of plasma HIV-1, it maintained a coincident relation to CSF HIV-1 in both viremic groups. In addition to correlation with CSF HIV-1 concentrations, CD8 activation in blood and CSF correlated with CSF WBCs and CSF neopterin.
Multivariate analysis confirmed the association of blood CD8 T-cell activation, along with plasma HIV-1 RNA and CSF neopterin, with CSF HIV-1 RNA levels.
Conclusions: The similarity of CD8 T-cell activation in blood and CSF suggests these cells move from blood to CSF with only minor changes in CD38/HLA-DR expression. Differences in the relation of CD8 activation to HIV-1 concentrations in the blood and CSF in the 2 viremic groups suggest that changes in immune activation not only modulate CSF HIV-1 replication but also contribute to CSF treatment effects. The magnitude of systemic HIV-1 infection and intrathecal macrophage activation are also important determinants of CSF HIV-1 RNA levels.
RESULTS
Subject Characteristics
The study included 123 HIV-1-infected (53 offs, 30 failures, and 40 successes) and 14 HIV-1-uninfected participants (Table 1). The subjects in the 4 groups were similar with respect to age, gender, and education. Among the infected subjects, group differences related to treatment history and were similar to those previously described for the larger cohort.13 The ongoing antiretroviral drug regimens of the 2 treatment groups were not different with respect to the number and class of combination therapies except for greater use of nonnucleoside reverse transcriptase inhibitors in the successes. Likewise, as in the larger cohort, the 2 treated groups did not differ in the number of CNS-penetrating drugs or in the duration of therapy (data not shown).13 None of the subjects suffered ongoing neurological disease.
Although the plasma HIV-1 RNA levels were not significantly different between the offs and failures (median 0.35 log10 copies/mL higher in offs than failures; Table 1), the CSF HIV-1 RNA levels were significantly higher in the offs than the failures (difference in medians of 1.65 log10, P < 0.001). This disproportionate treatment effect on CSF HIV-1 in the failures group was similar to that previously reported for the entire cohort at baseline.13 Also as previously reported, CSF WBC counts were elevated in the offs compared to the other 3 groups, which did not differ from each other. Similarly, CSF neopterin was higher in the offs, somewhat lower (though not significantly) in the failures, and lower still in the successes and HIV-negatives. There was no difference between the groups in blood:CSF albumin ratios.
Group Comparisons of T-Cell Activation
Figure 2 shows T-cell activation profiles in blood and CSF of the 4 subject groups. Compared to the offs (median 47.1%) (Fig. 2A), blood CD8 T-cell activation was clearly reduced in the failures (median 34.2%), with the successes showing further reduction (median 21.5%), yet not equal to the HIV-negative controls (median 9.2%), with overall comparison of groups by ANOVA showing P < 0.0001. Tukey's post hoc comparisons showed that the off group differed from the other 3 groups (P < 0.05 for failures, P < 0.001 for successes and offs); failures differed from successes (P < 0.01) and HIV-negatives (P < 0.001); and successes differed from HIV-negatives (P < 0.05). Although CD8 T-cell activation was somewhat higher in the CSF than in blood, this same comparative order was noted (Fig. 2B): medians for offs 61.3%, failures 39.1%, successes 26.7%, and HIV-negative controls 23.9%. In ANOVA analysis the overall comparison showed P < 0.0001, while post hoc comparisons showed that the offs group differed from the other 3 groups (P < 0.001 for all) and the failures differed from successes and HIV-negatives (P < 0.01), but successes did not differ from HIV-negatives. Hence, in both compartments, treatment had a clear effect on CD8 T-cell activation, and though the effect was greater in those with viral suppression, failed treatment also had a substantial impact. Overall, CSF CD8 T-cell activation correlated strongly with that of blood across the 4 groups (Fig. 2C), with a somewhat higher percentage of T cells isolated from CSF expressing activation markers (Y intercept in Figure 2C was 10.8% ± 2.0; median difference between CSF and blood values, 8.9%, IQR 2.7 to 16.4 across all groups).
FIGURE 2. Comparisons of CD8 and CD4 T-cell activation in blood and CSF of the 4 subject groups. A, Blood CD8 T-cell activation. Bars show medians and IQR. Highest activation was in the offs and lowest in HIV negatives. B, CSF CD8 cell activation showed similar order. C, CSF CD8 activation was highly correlated with that of blood when all groups were combined. The diagonal line shows the linear regression and 95% confidence intervals; the slope of regression was 0.95 with r2 = 0.72, P < 0.0001. D, Blood CD4 cell activation. E, CSF CD4 cell activation. F, Comparison of CSF to blood CD4 activation in all 4 groups showing regression line and 95% confidence intervals. Slope of regression is 0.67, with r2 = 0.26, P < 0.0001. Symbols designate subject groups as in C.
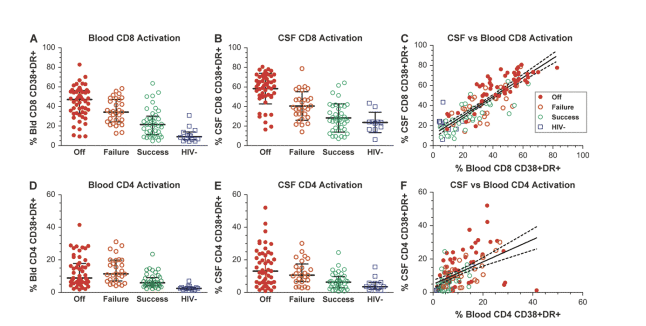
Though the CD4 T-cell activation in both blood and CSF (Fig. 2D and 2E) was higher in the infected subjects, there was not as clear a hierarchy as with CD8 T-cell activation (blood: offs, median 8.8%; failures, median 11.4%; successes, median 5.9%; CSF: offs, median 13.1%; failures, median 10.6%; successes, median 6.4%). For blood CD4 activation, ANOVA analysis showed an overall correlation with P < 0.0001. In post hoc comparisons, the offs group differed from successes (P < 0.01) and HIV-negatives (P < 0.001), but not from failures; failures differed from both successes (P < 0.01) and HIV-negatives (P < 0.001); and successes did not differ from HIV-negatives. For CSF, the overall comparison was significant (P < 0.0001), while post hoc comparisons found that offs differed from successes (P < 0.001) and HIV-negatives (P < 0.01), but not failures; the differences among the other 3 groups were not significant. Thus, although there was a proportional step from failures to successes to HIV negatives, as with CD8 activation, this was not seen between the offs and failures, which were nearly equal in both blood and CSF. The correlation between CSF and blood CD4 T-cell activation, though significant, was also not as robust as for CD8 T cells (Fig. 2F).
Both CSF and blood CD8 T-cell activation also correlated with the 2 other markers of CSF immune responses, CSF WBC counts (P < 0.0001; ρ = 0.450 and 0.380 for CSF and blood activation, respectively) and CSF neopterin (P < 0.0001; ρ = 0.647 and 0.637 for CSF and blood), as Figure 3 shows. In turn, CSF WBC count and CSF neopterin were also highly correlated (P < 0.0001; ρ = 0.407).
Relation of CD8 Activation to CSF HIV-1 in Offs and Failures
Because of distinct differences in CD8 activation between the offs and failures despite similar plasma viral loads, we extended our analysis to examine the associations of CD8 T-cell activation with viral loads in more detail and, more particularly, to compare the interactions of these variables in blood and CSF between the offs and failures. Figure 4 compares the relations of blood and CSF CD8 T-cell activation to HIV-1 RNA levels in both compartments in these 2 viremic groups. To analyze these relations, we used a model that included both the group effect (ie, failures versus offs) and the CD8 T-cell activation-HIV-1 RNA interaction effects. The results of this analysis showed that these relations were quite different with respect to plasma compared to CSF HIV-1 RNA levels. Thus, across the range of plasma RNA concentrations, blood and CSF CD8 T-cell activation levels were significantly lower in the failures than in the offs (Fig. 4A and 4B). In both compartments there was a downward shift in CD8 T-cell activation in the face of a given level of resistant HIV-1 compared to wild-type virus. The ANCOVA model used to analyze these relationships included both the group effect (ie, offs versus failures) and the group-by-viral-load interaction effect. In Figure 4A, blood CD8 T-cell activation was analyzed in relation to plasma HIV-1 RNA concentrations. The slopes for offs (5.59 ± 2.74; r2 = 0.088; P = 0.048) and failures (6.41 ± 3.09; r2 = 0.214; P = 0.010) were not different (P = 0.82), but their elevations were different (P = 0.001), indicating that across the plasma concentrations, CD8 T-cell activation was lower in the failures than in offs. In Figure 4B, CSF CD8 T-cell activation was analyzed in relation to plasma HIV-1 RNA. In the failures, increasing plasma HIV-1 RNA had little effect on CSF T-cell CD8 activation (P = 0.80), and the slopes in the offs (6.01 ± 2.69; r2 = 0.106; P = 0.031) and failures (0.80 ± 3.09; r2 = 0.002; P = 0.797) were not statistically different (P = 0.20); however, the elevations differed (P < 0.001).
FIGURE 4. Relation of blood and CSF CD8 T-cell activation to plasma and CSF HIV-1 RNA concentrations in the 2 viremic groups. The 4 panels compare the relations of CD8 T-cell activation to HIV-1 RNA concentrations in the offs (closed circles) and failures (open circles) in blood and CSF with regression lines (offs, solid line; failures, broken line) to emphasize the activation across the range of HIV-1 RNA concentrations. Because the lower range plasma HIV-1 RNA was defined in the failures at 500 copies/mL, values below this level in the offs were censored for this analysis and display; however, analysis that included these values and increased the slopes of the offs regression lines gave similar results (not shown).
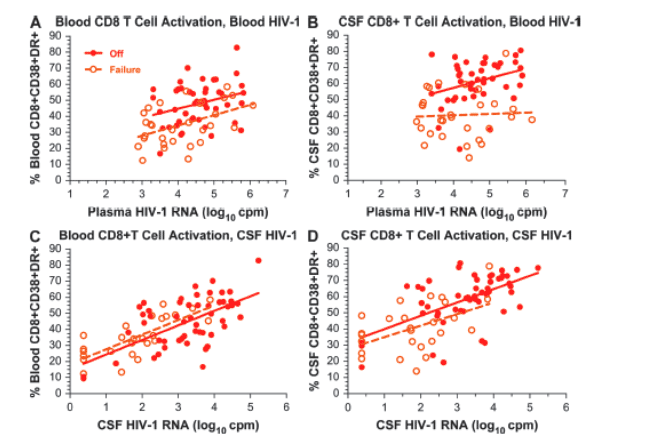
By contrast, there was no difference between the 2 viremic groups in the relation of either their blood or CSF CD8 T-cell activation to their CSF HIV-1 RNA concentrations (Fig. 4C and 4D). In Figure 4C, the blood CD8 T-cell activation was analyzed in relation to CSF HIV-1 RNA. Neither the slopes (P = 0.99) nor the elevations (P = 0.30) of the regression lines of the offs (9.08 ± 1.58; r2 = 0.393; P < 0.0001) differed from the failures (9.11 ± 1.57; r2 = 0.585; P < 0.0001), indicating that CSF had a coincident relation to activation across the range of CSF HIV-1 levels in both subject groups. In Figure 4D, CSF CD8 T-cell activation was analyzed in relation to CSF HIV-1 RNA. Again, neither the slopes (P = 0.72) nor the elevations (P = 0.09) differed in offs (slope = 8.22 ± 1.66; r2 = 0.333; P < 0.0001) and failures (slope = 7.13 ± 2.51; r2 = 0.252; P = 0.009). Hence, the failures and offs showed a similar level of CD8 T-cell activation in blood and CSF at a given level of CSF HIV-1. Because these relations between CD8 T-cell activation and CSF HIV-1 were not significantly different, the 2 treatment groups can be regarded as 1 population that showed a high correlation with CSF HIV-1 RNA for both blood and CSF CD8 T-cell activation (P < 0.0001 for both; blood activation r2 = 0.472 and CSF activation r2 = 0.440).
Multivariate Model of CSF HIV-1 Concentration
Univariate analysis found several of the measured variables to correlate highly (P < 0.0001) with the CSF HIV-1 RNA concentration across the 3 infected groups. These included blood (_ = 0.725) and CSF CD8 T-cell activation (_ = 0.758), plasma HIV-1 RNA (_ = 0.781), CSF neopterin (_ = 0.717), and CSF WBC count (_ = 0.546). Using the multiple regression procedure described in the Methods section, we entered each of these variables into the model to compare its associations with CSF HIV-1 RNA concentration across the 3 HIV-1-infected groups. The resultant final model was highly significant (P < 0.0001; r2 = 0.814) and included as significant predictors of CSF HIV RNA levels the variables plasma HIV-1 RNA (P < 0.001), CSF neopterin (P < 0.001), and blood CD8 activation (P = 0.015). CSF WBC count and CSF CD8 activation were not statistically significant at the 10% threshold and were therefore excluded from the final model; CSF WBC count was excluded after CSF neopterin was introduced and CSF CD8 activation after blood CD8 activation was introduced.
DISCUSSION
CSF provides an accessible and useful window into CNS infection and immune responses, albeit with the general caveat that brain and CSF responses are not always congruent.27 In this study, we demonstrate that flow cytometry can be used to analyze the activation state of CSF T cells, and that T-cell characterization affords novel insight into the pathogenesis and treatment of CSF HIV-1 infection. The 4 subject groups included in the study encompassed a diverse population with respect to HIV-1 infection, degree of immunosuppression, and treatment effect, allowing comparisons of blood and CSF cells across a broad range of CD8 and CD4 T-cell activation. The levels of activation of CSF CD8 cells and, to a lesser extent, CD4 cells were highly correlated with their blood counterparts across the subject groups and the range of cell activation. CSF CD8 T-cell coexpression of CD38 and HLA-DR was parallel to that in peripheral blood, though it was approximately 9% higher. This is consistent with the transmigration of these cells from blood to CSF with only minor selection or modification affecting these phenotypic characteristics, so that CSF T cells largely retain the activation profiles of those circulating in blood.
HIV-1 infection and treatment outcome had a marked effect on T-cell activation in both blood and CSF, with a stepwise decrease in the level of blood CD8 CD38/HLA-DR coexpression with partial and full viral suppression. Thus, although suppressive therapy had a greater impact, even failed treatment related to the development of drug resistance had a substantial and statistically significant effect, confirming previous reports related to blood cells19,28 and now extending this finding to CSF T cells. Comparison of activation in the 2 viremic groups in relation to plasma HIV-1 concentrations clearly showed that failed treatment lowered both blood and CSF CD8 activation compared to untreated subjects across the range of plasma viral loads (Fig. 4A and 4B) and that the reduced activation was not simply related to the (statistically insignificant) lower plasma HIV-1 RNA levels in the drug-resistant failures. The reason for this reduction in activation in relation to plasma viral load is not clear but has been suggested to relate in some way to reduced immunopathogenicity of drug-resistant virus.19 Whatever the mechanism, wild-type virus is associated with a higher level of activation than resistant virus at a given level of plasma viremia.
By contrast, a similar downward shift in activation in relation to CSF viral levels was not seen in failures compared to offs, so that the relation of blood and CSF CD8 T-cell activation to CSF viral load did not differ in the 2 viremic groups (Fig. 4C and 4D). Thus, there was a fundamental difference in the effects of virologic failure on the relation of activation to viral RNA levels in the 2 compartments. We favor the interpretation that the level of immune activation had a modulating effect on local CSF viral replication and that both resistant and wild-type viruses replicated similarly at a given level of immune activation in this compartment. Thus, although in the blood, activation depends on whether infection is caused by wild-type or resistant virus, in the CSF this relation does not hold; rather, activation determines a similar level of replication for both types of virus. The reasons for this are not clear, but may relate to the fact that systemic virus originates in lymphoid tissue, whereas CSF virus relies on transmigration of lymphocytes and monocytes into a nonlymphoid organ. Cell activation may be an important determinant of this transmigration. Whatever the mechanism, this relation between activation and CSF HIV-1 RNA may explain some of the effects of antiretroviral treatment on CSF HIV-1 RNA concentrations, including the disproportionate effect of failed treatment reported earlier in this cohort13 and noted again for the study visits reported here, with CSF HIV-1 RNA levels more than 10-fold lower in the failures than the offs, despite nearly equal plasma HIV-1 RNA levels (Table 1).
The multivariate analysis of the possible contributions to CSF HIV-1 RNA concentrations suggested that the measured variables could be segregated into 3 groups: the plasma HIV-1 RNA concentration; the CD8 T-cell activation, with the blood CD8 activation occluding the effect of CSF CD8 activation (when blood CD activation was added to the model, CSF activation was no longer significant); and the CSF neopterin and WBC counts, with the former occluding the effect of the latter (addition of CSF neopterin to the model eliminated the WBC count as a significant contributor). These variables are clearly intimately interrelated, both statistically and biologically.
The combination of our earlier observations13 with these findings related to CD8 T-cell activation can be synthesized into a coherent set of observations and derivative pathogenic hypothesis as follows: First, systemic HIV-1 infection drives generalized immune activation29 that includes both CD8 and CD4 T cells and, likely, monocyte/macrophages.30 Second, CD8 T-cell activation, whether measured in blood or CSF, serves as a useful index of this generalized immune activation.21,31 Third, resistant virus is a less potent driver of systemic activation than wild-type virus. Fourth, by virtue of their origin from blood, CSF T cells retain their activated blood phenotype, which is determined by the level of systemic immune activation rather than local factors; indeed, statistically, blood CD8 activation more directly accounts for the contribution of T-cell activation than CSF CD8 activation. Fifth, HIV-1 replication of both wild-type and resistant viruses in the CSF is similarly modulated by the level of this local immune activation. Sixth, although the mechanism of this modulation is uncertain, it may involve the increased availability of target CD4 T cells due to their increased cell traffic (an idea supported by the correlation between CD8 activation and CSF pleocytosis) and to their higher capacity for supporting viral replication as a result of their activated state. Thus, activation state might contribute to CSF viral replication and local viral amplification by at least 2 mechanisms: an effect on cell migration (cell availability) and an effect on viral output from the local cells (viral production). Other major contributors to CSF HIV-1 concentration include the magnitude of systemic infection, as indicated by the plasma HIV-1 RNA concentration, and intrathecal macrophage activation, indicated by CSF neopterin.
This reasoning, which underlies the simple model of CSF HIV-1 infection diagrammed in Figure 5, suggests that the bulk of the virus detected in CSF derives from amplified infection that is modulated by the level of immune activation. The simple schematic in the figure depicts the principal compartments participating in the origin of CSF infection-systemic (blood), leptomeninges (CSF), and brain-and the hypothesized role of immune activation in transitory, autonomous, and amplified CSF infection. In the systemic compartment, HIV-1 replication leads to blood viremia and stimulates generalized immunoactivation, including systemic T-cell activation, which can be measured using CD8 CD38 and HLA-DR surface coexpression as an index. Although this activation modulates HIV-1 replication, systemic HIV-1 replication is the prime mover in this interaction. In the leptomeninges compartment, systemic viremia (especially CD4 T-cell-associated infection) is responsible for initial seeding. This direct hematogenous seeding is responsible for transitory CSF infection that predominates during primary HIV-1 infection. In the brain compartment, chronic brain infection sustained within longer-lived perivascular and parenchymal macrophages and microglial cells (together encompassed by the abbreviation M_), termed autonomous infection, can also spill over into the CSF. Macrophages presumably also participate in the generalized immune activation caused by HIV-1, as suggested by the elevated CSF neopterin and its correlation with CD8 T-cell activation in CSF and blood. The multivariate analysis suggests that macrophage activation contributes to CSF pleocytosis as well. Activated CSF T cells originate in the blood and importantly contribute to local intrathecal (and perhaps perivascular) amplified infection by supplying susceptible CD4 T cells that sustain local replication within the CSF space. This amplified infection may be continuously reseeded from the blood or from the brain sources (ie, from transitory or autonomous mechanisms) and modified by local viral evolution and selection. The level of CD4 T-cell activation is a major factor in sustaining the level of this local amplification. Though measurements of CD4 T cells do not correlate as well with CSF HIV-1 levels as CD8 activation does, this may be explained by direct infection of the former and their preferential destruction.
Responses of CSF infection to treatment may depend on both direct and indirect effects. The chief direct target may be systemic infection, reflected in the plasma HIV-1 response, whereas direct effects may be weaker behind the BCB and BBB because of reduced drug penetration. However, targeting systemic infection will secondarily reduce CSF infection by several mechanisms, including reduction of new transitory seeding as both the influx of hematogenously circulating infected cells and their virus output are reduced. Reduction of systemic infection will also reduce systemic T-cell activation. Because these cells are the source of activated CSF T cells, they will also be reduced in the CSF and in turn will lead to lower amplification. Effects on macrophage activation may also reduce autonomous infection and even amplified infection through modulation of T-cell chemotaxis.
This model is also consistent with observations of genetically compartmentalized infection32,33 and with suggestions that CSF HIV-1 derives principally from short-lived cells, likely lymphocytes.32 When applied to understanding treatment effects, this model suggests that reducing systemic immune activation with ART may contribute to suppressing CSF infection, not only in the failures but also in the successes. Because of the BBB and BCB, most antiviral drugs do not achieve concentrations in brain or CSF comparable to those in plasma or systemic organs and therefore might be expected to be less effective in these compartments.34 However, as in this group of subjects, most studies of treatment on CSF HIV-1 have shown effective viral suppression.5,14-16 Although in some patients CSF HIV-1 RNA decays with treatment more slowly than plasma RNA does, in many CSF virus falls just as rapidly through the first phase of decay, and good overall therapeutic responses seem to be the rule with occasional notable exceptions.5,35-37 How is this possible, with CSF's lower drug access? Reduction of immune activation, leading to both reduced influx of infected cells and down-regulation of local amplification, may contribute importantly. Autonomous infection within meningeal and perivascular macrophages and microglial cells38 likely is also modulated as part of the generalized immunoactivation, as suggested by the correlation of CD8 activation with CSF neopterin (a marker of local macrophage activation and intrathecal immunoactivation25,39) and the contribution of CSF neopterin to the multivariate model of CSF HIV-1 RNA concentrations. Indeed, local macrophage activation, as indicated by neopterin concentrations, likely also contributes to CSF pleocytosis, perhaps through chemotactic signals or other mechanisms.
CNS HIV-1 infection is an important component of systemic infection and a critical target of therapy. These studies suggest that the level of systemic immune activation is an important modulator of this infection and that its downregulation by ART may contribute to controlling HIV-1 in this compartment and explain the better-than-predicted responses of CSF HIV-1 to ART and both the preventative40 and therapeutic41 effects of treatment on ADC, although importantly the current study was confined to subjects without neurological impairment.
|
|
| |
| |
|
|
|