| |
Raltegravir with Optimized Background Therapy for Resistant HIV-1 Infection
|
| |
| |
NEJM July 24, 2008
Roy T. Steigbigel, M.D., David A. Cooper, M.D., D.Sc., Princy N. Kumar, M.D., Joseph E. Eron, M.D., Mauro Schechter, M.D., Ph.D., Martin Markowitz, M.D., Mona R. Loutfy, M.D., M.P.H., Jeffrey L. Lennox, M.D., Jose M. Gatell, M.D., Ph.D., Jurgen K. Rockstroh, M.D., Christine Katlama, M.D., Patrick Yeni, M.D., Adriano Lazzarin, M.D., Bonaventura Clotet, M.D., Jing Zhao, Ph.D., Joshua Chen, Ph.D., Desmond M. Ryan, B.S., Rand R. Rhodes, M.S., John A. Killar, M.S., Lucinda R. Gilde, B.S., Kim M. Strohmaier, B.S., Anne R. Meibohm, Ph.D., Michael D. Miller, Ph.D., Daria J. Hazuda, Ph.D., Michael L. Nessly, M.S., Mark J. DiNubile, M.D., Robin D. Isaacs, M.D., Bach-Yen Nguyen, M.D., Hedy Teppler, M.D., for the BENCHMRK Study Teams
ABSTRACT
Background Raltegravir (MK-0518) is an inhibitor of human immunodeficiency virus type 1 (HIV-1) integrase active against HIV-1 susceptible or resistant to older antiretroviral drugs.
Methods We conducted two identical trials in different geographic regions to evaluate the safety and efficacy of raltegravir, as compared with placebo, in combination with optimized background therapy, in patients infected with HIV-1 that has triple-class drug resistance in whom antiretroviral therapy had failed. Patients were randomly assigned to raltegravir or placebo in a 2:1 ratio.
Results In the combined studies, 699 of 703 randomized patients (462 and 237 in the raltegravir and placebo groups, respectively) received the study drug. Seventeen of the 699 patients (2.4%) discontinued the study before week 16. Discontinuation was related to the study treatment in 13 of these 17 patients: 7 of the 462 raltegravir recipients (1.5%) and 6 of the 237 placebo recipients (2.5%). The results of the two studies were consistent. At week 16, counting noncompletion as treatment failure, 355 of 458 raltegravir recipients (77.5%) had HIV-1 RNA levels below 400 copies per milliliter, as compared with 99 of 236 placebo recipients (41.9%, P<0.001). Suppression of HIV-1 RNA to a level below 50 copies per milliliter was achieved at week 16 in 61.8% of the raltegravir recipients, as compared with 34.7% of placebo recipients, and at week 48 in 62.1% as compared with 32.9% (P<0.001 for both comparisons). Without adjustment for the length of follow-up, cancers were detected in 3.5% of raltegravir recipients and in 1.7% of placebo recipients. The overall frequencies of drug-related adverse events were similar in the raltegravir and placebo groups.
Conclusions In HIV-infected patients with limited treatment options, raltegravir plus optimized background therapy provided better viral suppression than optimized background therapy alone for at least 48 weeks.
Highly active antiretroviral therapy is the standard of care for patients with advanced human immunodeficiency virus (HIV) infection.1 Combination regimens have resulted in improved survival, decreased morbidity, and cost-effective care for patients with a CD4 count of less than 350 per cubic millimeter.2,3,4,5,6,7,8 However, viral suppression cannot always be achieved or sustained with standard treatments because of the development of viral resistance, toxic effects of drugs, or lack of adherence.9,10,11,12,13,14,15,16,17,18 The majority of HIV-infected patients in whom highly active antiretroviral therapy fails have resistant viral quasispecies.12,13,14,15,19,20 Cross-resistance to agents within a drug class may exhaust most available treatment options.1,21 Antiretroviral drugs directed at new HIV targets are urgently needed for patients with unsuppressed viremia despite treatment.14,22,23,24
Integrase is a viral enzyme that is essential for HIV type 1 (HIV-1) replication, catalyzing the insertion of proviral DNA into the host-cell genome.25 Because HIV-1 integrase represents a distinct therapeutic target, integrase inhibitors would be expected to maintain activity against HIV-1 strains even when resistant to other classes of antiretroviral drugs.26,27,28 Raltegravir (MK-0518; Isentress, Merck) specifically inhibits proviral DNA-strand transfer, with potent in vitro activity against HIV-1 that is susceptible or resistant to other classes of antiretroviral drugs.27,29,30 In combination with optimized background therapy in a placebo-controlled, dose-ranging phase 2 study involving patients with resistant infection, raltegravir given at a dose of 200 mg, 400 mg, or 600 mg twice daily suppressed plasma HIV-1 RNA to a level below 50 copies per milliliter at week 24 in 56 to 67% of recipients across the dose groups, as compared with 13% of patients receiving optimized background therapy alone.31 Because the three doses tested in the phase 2 program were similarly efficacious and had acceptable side-effect profiles, the middle dose of 400 mg twice daily was chosen for further study, providing a margin of safety and efficacy when raltegravir is coadministered with drugs that are inhibitors or inducers of its major metabolizing enzyme (uridine diphosphate glucuronosyltransferase isoform 1A1).29 In this report, we present clinical trial data on the safety, adverse-effect profile, and virologic and immunologic effects of combining raltegravir (400 mg twice daily) with optimized background therapy in HIV-infected adults with unsuppressed viremia despite antiretroviral therapy.
Methods
The studies were designed, managed, and analyzed by the sponsor in conjunction with the academic authors. The authors had access to all study data on request. This report was principally drafted by two academic authors and four industry authors and was critically reviewed and approved by all the authors in its final form before submission. All authors vouch for the completeness and accuracy of the data.
Study Design
There are two ongoing BENCHMRK (Blocking Integrase in Treatment Experienced Patients with a Novel Compound against HIV, Merck) studies. BENCHMRK-1 (MK-0518 protocol 018, Merck) is a double-blind (including blinding of the sponsor), randomized, phase 3 clinical trial being conducted in Europe, Asia, Australia, and Peru. BENCHMRK-2 (MK-0518 protocol 019, Merck) in North and South America is identical in design to the concurrent BENCHMRK-1 trial. The protocol was approved by the institutional review board or ethics review committee at each site. All participants provided written informed consent. The planned total duration of each study is at least 156 weeks of the study therapy. This report presents efficacy results through week 48 and all available safety data from the groups through August 3, 2007, for BENCHMRK-1 and July 31, 2007, for BENCHMRK-2 (the dates when the last patient remaining in the double-blind phase of the study completed the 48-week visit).
Patients
HIV-infected patients 16 years of age or older were eligible if they had HIV-1 RNA levels of more than 1000 copies per milliliter while receiving antiretroviral therapy, with documented phenotypic or genotypic resistance to at least one drug in each of three classes of oral antiretroviral drugs (nucleoside reverse-transcriptase inhibitors, non-nucleoside reverse-transcriptase inhibitors, and protease inhibitors). Exclusion criteria were renal insufficiency (a serum creatinine level greater than twice the upper limit of the normal range), acute or decompensated chronic hepatitis, uncontrolled substance abuse, and any medical condition likely to interfere with the execution or interpretation of the study. Pregnant or breast-feeding women were also excluded. Patients with stable chronic hepatitis B or hepatitis C were eligible if their serum aminotransferase levels were less than five times the upper limit of the normal range. Patients with active cancer could be enrolled if chemotherapy was not required and the cancer was not considered likely to obscure the study outcome.
Study Treatment
At entry, investigators selected optimized background therapy for each patient on the basis of the history of antiretroviral treatment, results of available drug-resistance tests, and previous or current laboratory data. Darunavir and tipranavir, which were investigational at the time of the studies, were permitted as part of optimized background therapy, subject to local regulatory approval, to ensure the most active regimen for patients. The study drug and optimized background therapy were initiated concurrently. Subsequent changes to optimized background therapy were permitted only for management of toxic effects or if patients switched to open-label raltegravir because of virologic failure after week 16.
Patients were randomly assigned to raltegravir or placebo in addition to their optimized background therapy, in a 2:1 ratio, through a central, interactive voice-response system, according to a computer-generated, randomized allocation schedule. Randomization was stratified according to both the use or nonuse of enfuvirtide in the optimized background therapy and the degree of viral resistance to protease inhibitors (resistance to 1 vs. >1). Investigators, study site and sponsor personnel, patients, and laboratory personnel remained unaware of the treatment assignments and dose, with blinding accomplished through the use of placebo tablets that were identical in appearance to the raltegravir tablets. Participants were instructed to take one 400-mg tablet of raltegravir or placebo twice daily, 12 hours (±2 hours) apart, without regard to food intake.
Assessments
Clinical status was assessed at regularly scheduled visits and as needed; protocol-mandated laboratory tests were performed in a central laboratory. HIV-1 RNA levels were measured with the standard polymerase-chain-reaction (PCR)assay (Cobas Amplicor HIV-1 Monitor assay, version 1.5; Roche Diagnostics), with lower and upper limits of quantification of 400 and 750,000 copies per milliliter, respectively. All samples for which the RNA levels were below the lower limit were evaluated with the use of a more sensitive PCR assay (Ultrasensitive Amplicor HIV-1 Monitor assay, version 1.5; Roche Diagnostics), with a lower quantification limit of 50 copies per milliliter. Resistance testing (PhenoSense GT, Monogram Biosciences), was mandated at baseline and at the time of virologic failure.
Sensitivity Scores
Genotypic and phenotypic sensitivity scores for the optimized background therapy were determined from the results of baseline resistance tests. The genotypic and phenotypic sensitivity scores were the total number of antiretroviral drugs used as part of the optimized background therapy to which a patient's HIV was fully susceptible, as determined with the use of genotypic and phenotypic resistance testing, respectively. For the calculation of genotypic sensitivity scores, the drug-susceptibility results were interpreted on the basis of the Monogram algorithm available at the time. For the calculation of phenotypic sensitivity scores from the PhenoSense GT results, only antiretroviral drugs in the optimized background therapy that were active at concentrations below the cutoff point established for the fully active drug were considered to be active. A score of 0 does not rule out reduced but clinically meaningful antiretroviral activity for drugs included in the optimized background therapy. Because resistance criteria for darunavir and enfuvirtide were not available from the PhenoSense GT analysis during the study, each of the two drugs was counted (+1) in both phenotypic and genotypic sensitivity scores when used as part of the optimized background therapy in patients who had not previously been given that agent. Low-dose ritonavir (<600 mg per day) was not counted as a separate drug.
Virologic Failure
At or after week 16, virologic failure was considered to have occurred if the patients did not have a decrease in the HIV-1 RNA level to less than 400 copies per milliliter or by more than 1 log10 copies per milliliter from the baseline level; if they had an increase in the HIV-1 RNA level of more than 1 log10 copies per milliliter from the nadir level on two consecutive measurements; or if they had two consecutive HIV-1 RNA measurements of 400 or more copies per milliliter after having had a level of less than 400 copies per milliliter. Patients with virologic failure could opt to remain in the blinded portion of the study, enter an open-label phase and receive raltegravir as part of a new regimen, or withdraw from the study.
Adjudication of Events and Oversight by the Data and Safety Monitoring Board
Events occurring during the studies that were suspected to indicate the presence of the acquired immunodeficiency syndrome (AIDS) (other than a CD4 cell count 200 cells per cubic millimeter) were reviewed by an independent adjudicator with expertise in HIV medicine who was unaware of the treatment assignment.
An independent data and safety monitoring board periodically reviewed the blinded safety and efficacy results and could request unblinded data and make nonbinding recommendations regarding the completion of enrollment and continuation of the studies. As planned, the data and safety monitoring board also reviewed the unblinded results of the primary analysis after all participants remaining in the double-blind phases of the studies had completed at least 16 weeks of follow-up (at which point the median duration of follow-up was 26 weeks [range, 1 to 39] in the raltegravir groups and 21 weeks [range, 4 to 40] in the placebo groups).
Statistical Analysis
The prespecified primary efficacy hypothesis for each BENCHMRK study was that, when used in combination with optimized background therapy, raltegravir would have superior antiretroviral activity to that of placebo, based on the proportion of patients with HIV-1 RNA levels of less than 400 copies per milliliter after 16 weeks of study therapy. Assuming a true response rate at week 16 of 70% among raltegravir recipients and 50% among placebo recipients and anticipating a 10% rate of discontinuation for reasons not related to treatment, we estimated that a study with a planned enrollment of 230 patients in the raltegravir group and 115 patients in the placebo group would have a statistical power of 90% to show superiority of optimized background therapy with raltegravir over optimized background therapy with placebo. Additional prespecified efficacy outcomes included the proportions of patients with HIV-1 RNA levels of less than 50 copies per milliliter and the change from baseline in CD4 cell counts.
All treated patients were included in the efficacy and safety analyses. Missing virologic data were handled in three prespecified ways.31 The primary analysis used a treatment-related discontinuation approach that considered patients who discontinued the double-blind phase of the study owing to lack of efficacy or adverse events (or whose HIV-1 RNA level was 400 [or 50] copies per milliliter at the time of discontinuation for reasons not related to treatment) as having treatment failure at subsequent time points. HIV-1 RNA measurements that were missing because of skipped or mistimed visits were left as missing. A second, worst-case approach considered noncompletion as treatment failure at subsequent time points. HIV-1 RNA measurements that were missing because of skipped or mistimed visits were imputed as treatment failures in the noncompletion analysis unless the values immediately before and after the missing value both indicated successful treatment, in which case the missing value was left as missing. A third, observed-failure approach considered discontinuation due to lack of efficacy as treatment failure at subsequent time points. No other missing values were imputed.
A logistic-regression model was used to compare virologic-response rates between the two treatment groups, after adjustment for covariates that might affect the likelihood of achieving HIV RNA suppression. Independent variables incorporated into the model were the HIV-1 RNA level at baseline, presence or absence of an active protease inhibitor in the optimized background therapy (as determined by phenotypic resistance testing), first use of darunavir in the optimized background therapy (vs. either use in patients previously treated with darunavir or nonuse), first use of enfuvirtide in the optimized background therapy (vs. either use in patients previously treated with enfuvirtide or nonuse), and the study drug.
Combined analyses of both BENCHMRK studies were prespecified to provide precise estimates of treatment effects. For the combined analysis of the proportion of patients who had HIV-1 RNA levels of less than 400 copies per milliliter at week 16, a covariate for the BENCHMRK study and an interaction term between study drug and BENCHMRK study were included in the logistic-regression model to examine the homogeneity of the treatment effects between the two BENCHMRK studies.
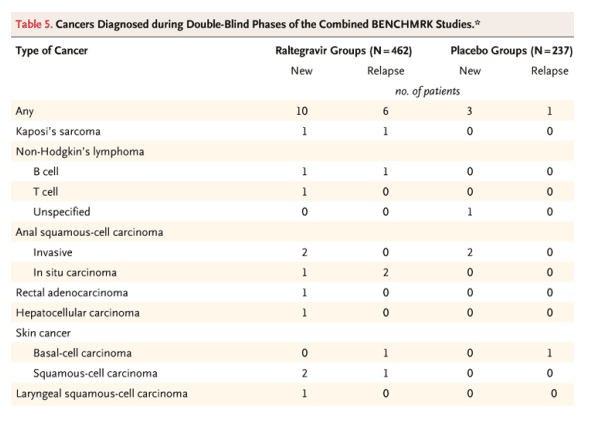
Adverse events occurring during the double-blind phase of the study or within 14 days after discontinuation of blinded therapy were tabulated until the cutoff dates for each analysis. Investigators, while unaware of treatment assignment, were to assess the relationship of each adverse event to the study therapy; adverse events were counted as drug-related if they were judged by the investigator as definitely, probably, or possibly related to any of the study drugs (including drugs used in the optimized background therapy). The severity of laboratory abnormalities was graded according to the 1992 Division of Acquired Immunodeficiency Syndrome (DAIDS) toxicity criteria for adults (http://rcc.tech-res-intl.com/tox_tables.htm). The frequencies of adverse events were not adjusted for duration of follow-up except for calculations of relative risk. Diagnoses of new, progressive, or recurrent cancers, regardless of histologic grade and including in situ anal carcinoma and nonmelanoma skin cancer, were tallied as cancers. Incidence, relative risk, and time to diagnosis of cancer were based solely on the first cancer diagnosed during the double-blind phase.
All statistical tests were two-sided. P values of less than 0.05 were considered to indicate statistical significance, except for the interaction between study drug and BENCHMRK study, for which P values of less than 0.10 were considered to indicate statistical significance.
Results
Enrollment, Follow-up, and Baseline Characteristics of Patients
Figure 1 in Supplementary Appendix 2 (available with the full text of this article at www.nejm.org) summarizes the enrollment and follow-up of patients until the cutoff dates of August 3, 2007, for BENCHMRK-1 and July 31, 2007, for BENCHMRK-2. Because discontinuations for virologic failure and entry into the open-label phase occurred more frequently among placebo recipients than among raltegravir recipients, the median duration of follow-up for the safety analysis during the double-blind phase was 57.4 weeks (range, 3.0 to 72.0) in the raltegravir groups and 37.6 weeks (range, 5.6 to 72.9) in the placebo groups for the combined BENCHMRK studies.
Baseline characteristics were generally balanced between the two treatment groups within each BENCHMRK study (Table 1). The majority of enrolled patients were heavily pretreated white men with AIDS. Overall, 12.0% of participants were women and 32.2% of participants were nonwhite. Before enrollment, 46.2% and 7.3% of patients had received enfuvirtide and darunavir, respectively. At entry, 16.2% of patients had chronic hepatitis B or hepatitis C coinfection and 20.0% had a history of cancer or a premalignant condition.
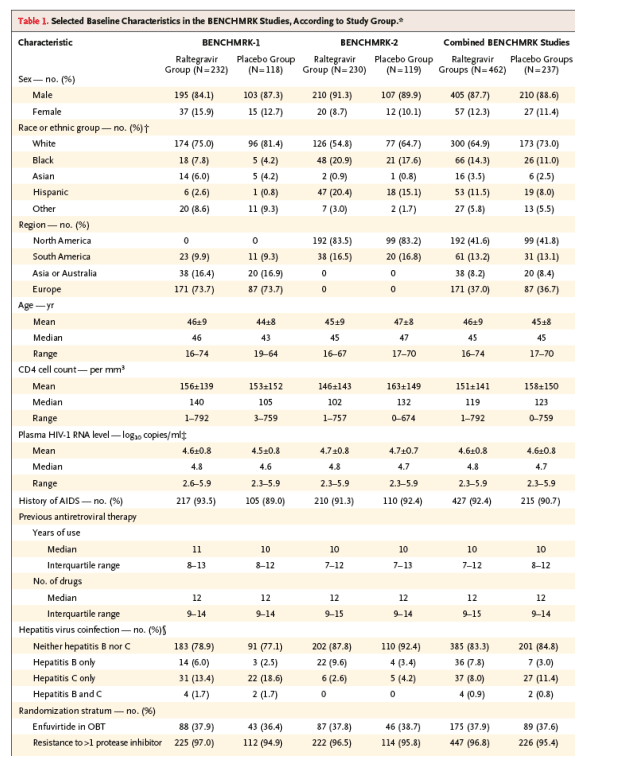
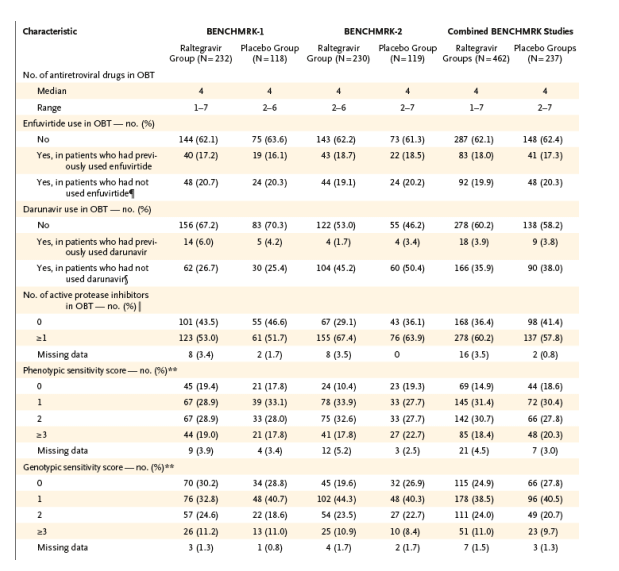
Virologic and Immunologic Responses
Since few patients discontinued either BENCHMRK study prematurely, the results of the three statistical approaches for handling missing data were similar (Table 2). The treatment effect was consistent between the two BENCHMRK studies (P>0.10 for homogeneity with each approach). On the basis of the primary, prespecified analysis counting treatment-related discontinuation as failure, HIV-1 RNA levels below 400 copies per milliliter at the primary (week 16) time point were achieved in 178 of 227 raltegravir recipients (78.4%) as compared with 48 of 117 placebo recipients (41.0%) in BENCHMRK-1 (P<0.001) and in 177 of 226 raltegravir recipients (78.3%) as compared with 51 of 118 placebo recipients (43.2%) in BENCHMRK-2 (P<0.001). According to the more stringent analysis counting any noncompletion as treatment failure, HIV RNA levels were reduced to less than 400 copies per milliliter at week 16 in 355 of 458 raltegravir recipients (77.5%) as compared with 99 of 236 placebo recipients (41.9%) (P<0.001 for each study individually and the combined studies) (Figure 1). HIV-1 RNA was suppressed to a level of less than 50 copies per milliliter at 16 weeks in 61.8% of the raltegravir group as compared with 34.7% of the placebo group (P<0.001 for each study individually and the combined studies).
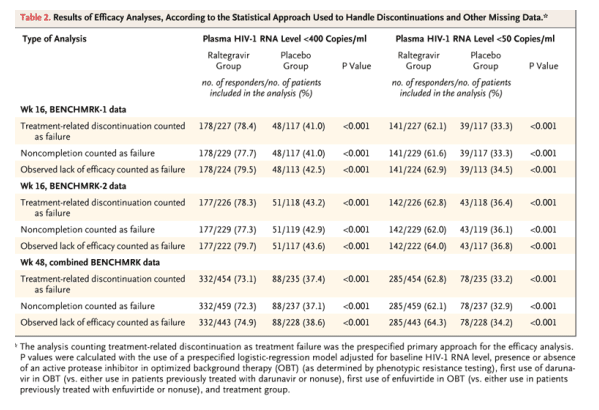
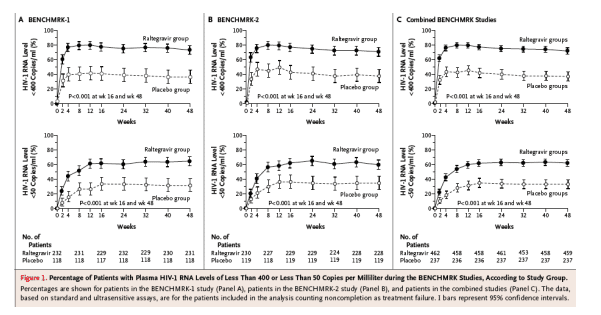
In the combined analysis considering noncompletion as failure at week 48, HIV-1 RNA levels were reduced to less than 400 copies per milliliter in 332 of 459 raltegravir recipients (72.3%) as compared with 88 of 237 placebo recipients (37.1%) (P<0.001 for each study individually and the combined studies). In the combined analysis, HIV-1 RNA levels were suppressed to less than 50 copies per milliliter at week 48 in 285 of 459 raltegravir recipients (62.1%) as compared with 78 of 237 placebo recipients (32.9%) (P<0.001 for each study individually and the combined studies).
On the basis of the approach considering observed lack of efficacy as treatment failure, the overall mean change in log10 HIV-1 RNA copies per milliliter at week 48 was -1.7 (95% confidence interval [CI], -1.8 to -1.6) in the raltegravir groups and -0.8 (95% CI, -0.9 to -0.6) in the placebo groups (P<0.001 for both studies individually and combined) (Figure 2 in Supplementary Appendix 2). The mean change in CD4 cell counts between baseline and week 48 was 109 per cubic millimeter (95% CI, 98 to 121) in the raltegravir groups as compared with 45 per cubic millimeter (95% CI, 32 to 57) in the placebo groups (P<0.001 for each study individually and the combined studies).
Confirmed AIDS-defining clinical events occurred by week 48 in 17 of the 462 patients (3.7%) in the raltegravir groups and 11 of the 237 patients (4.6%) in the placebo groups (Table A1 in Supplementary Appendix 2). In comparisons of the numbers of patients with AIDS-defining conditions in the raltegravir groups and the placebo groups, based on the person-years of follow-up in the double-blind phases of both studies, the relative risk of an AIDS-defining event with raltegravir was 0.59 (95% CI, 0.26 to 1.40). The median time at which an AIDS-defining event was diagnosed was 64 days (interquartile range, 52 to 110) among raltegravir recipients and 105 days (interquartile range, 19 to 118) among placebo recipients.
Safety, Adverse Events, and Side Effects
During the double-blind phases from both BENCHMRK studies, clinical adverse events of any intensity were reported in 90.3% of patients in the raltegravir groups and 88.2% of patients in the placebo groups and were considered to be related to any drug in the study regimen in 54.8% of patients receiving raltegravir and 55.3% of those receiving placebo; laboratory adverse events occurred in 25.5% of raltegravir recipients and 23.2% of placebo recipients and were considered to be drug-related in 14.7% and 13.5%, respectively (Table 3). Other than local reactions at the site of enfuvirtide injections, the most common drug-related clinical adverse events in both treatment groups were diarrhea, nausea, and headache. The most common drug-related laboratory adverse events were increased serum cholesterol, triglyceride, and aminotransferase levels in the raltegravir groups and increased cholesterol and creatinine levels and decreased neutrophil counts in the placebo groups. Moderate-to-severe clinical adverse events and laboratory abnormalities of grade 3 or 4 are reported in Table 4. Supplementary Appendix 2 lists all serious clinical adverse events (Table A2), clinical adverse events of any intensity or causality that occurred in 2% or more of patients in either treatment group (Table A3), and laboratory abnormalities of DAIDS grade 2, 3, or 4 (Table A4).
Discussion
Highly active antiretroviral therapy effectively suppresses viremia in most patients with advanced HIV infection who comply with their antiretroviral regimen, but drug-resistant virus emerges in some patients.12,13,14,20,32,33,34 In addition, transmission of resistant HIV places patients who have not previously received treatment at risk for suboptimal responses to first-line regimens.10,15,16,17,18,35,36 Historically, as compared with the initial regimens, subsequent antiretroviral regimens used in patients with resistant virus after treatment failures have been much less successful in suppressing viremia.1,12,20,21,32,33,37,38 Raltegravir inhibits HIV-1 replication through a mechanism of action distinct from that of other currently approved antiretroviral drugs and thus offers the promise of potent activity against multidrug-resistant virus.26,27,28,31
In patients infected with HIV that had triple-class drug resistance, raltegravir given at a dose of 400 mg twice daily at intervals of 12 (±2) hours, with optimized background therapy, had a rapid and potent antiretroviral effect that was superior to the effect of optimized background therapy alone at week 48. These results were consistent between the two BENCHMRK studies. The majority of patients in both studies had AIDS at entry and had failure of multiple previous antiretroviral regimens, with the emergence of highly resistant virus. The inferior virologic responses in the placebo groups as compared with the raltegravir groups were consistent with the findings in other studies targeting similar populations and attest to the shortcomings of older treatment options in this context.39,40,41,42,43,44,45,46,47 Immunologic benefits evidenced by increased CD4 cell counts were also observed during raltegravir therapy.
Overall, adverse-event profiles were generally similar for the raltegravir and placebo regimens. The disproportionate diagnosis of several cancers in the raltegravir groups as compared with the placebo groups at the time of the 16-week analyses in the two BENCHMRK studies prompted a comprehensive review of all cancers occurring in the four phase 2-3 trials of raltegravir.31,48,49 Data from three studies of heavily pretreated patients were combined with data from one study of previously untreated patients and were adjusted for the duration of follow-up during the double-blind phases.50 The relative risk of cancer associated with raltegravir, as compared with placebo, was 1.2 (95% CI, 0.4 to 4.1) in the most current analysis, with a composite rate of 2.2 cancers per 100 patient-years in the raltegravir groups (vs. 1.8 per 100 patient-years in the placebo groups). The types and frequencies of cancers were similar to those reported in patients with advanced HIV infection.51 Most patients with a diagnosis of cancer had favorable virologic and immunologic responses. Cancers were generally detected earlier after the initiation of study therapy in the raltegravir groups than in the placebo groups. Although usually associated with AIDS-related opportunistic infections, the immune reconstitution syndrome has been linked to the detection of cancer.52,53,54,55,56,57,58,59,60,61,62 No evidence of carcinogenicity was apparent from the preclinical data on raltegravir. Continued vigilance in the monitoring of safety is nonetheless warranted.
Our results should be interpreted in the context of the prelicensure experience with this new class of drugs in a limited number of patients to date.31,48,49 Data on the efficacy, safety, and tolerability of raltegravir given for 48 or more weeks are accumulating.49,63 The durability of viral suppression and the tolerability of longer-term therapy with raltegravir-based combination regimens continue to be assessed in the ongoing BENCHMRK studies. The development of resistance to integrase inhibitors is a particular concern when raltegravir is by necessity used as functional monotherapy (in which there are no fully active drugs in the optimized background therapy).12,20,48,64,65 Although viral suppression has been found in some patients whose optimized background therapy had a genotypic or phenotypic sensitivity score of 0, some drugs used in the optimized background therapy may have retained partial antiretroviral activity.48 In contrast, baseline resistance testing may have failed to detect archived resistance mutations present at frequencies below the limit of detection of the assay, thereby possibly inflating some sensitivity scores in both treatment groups.
In patients with few remaining treatment options, raltegravir combined with optimized background therapy provided superior HIV-1 suppression as compared with optimized background therapy alone, despite the presence of virus that has triple-class drug resistance. The potent suppression of viremia seen at week 16 in raltegravir recipients was usually achieved by week 4 and sustained through week 48. As with other new antiretroviral agents under development that work through novel mechanisms of action,66 raltegravir as part of an optimized combination regimen offers hope for the suppression of HIV-1 infection that is resistant to standard therapies.67,68
Supported by Merck.
|
|
| |
| |
|
|
|