 |
|
|
| |
The pharmacokinetic (PK) profile of darunavir with low-dose ritonavir (DRV/r) in various multiple-dose regimens over 120 hours
|
| |
| |
Reported by Jules Levin
9th International Workshop on Clinical Pharmacology of HIV Therapy
April 7-9, 2008
New Orleans
Marta Boffito,1 Graeme Moyle,1 Andrew Hill,2,3 Vanitha Sekar,4 Eric Lefebvre,5 Martine De Pauw,3 Ralph DeMasi,4 Richard Hoetelmans3
1Pharmacokinetic Research Unit, St Stephen's Centre, Chelsea and Westminster Hospital, London, UK; 2Department of Pharmacology, University of Liverpool, Liverpool, UK;
3Tibotec BVBA, Mechelen, Belgium; 4Tibotec Inc., Yardley, PA, USA; 5Janssen-Cilag, Tilburg, The Netherlands
AUTHOR CONCLUSIONS
Following repeated DRV/r dosing of 800/100mg once-daily, DRV concentrations
remained above the EC50 value of 55ng/mL for at least 48 hours in all volunteers after the last administered DRV dose while ritonavir dosing was continued.
Once-daily DRV/r administered at a dose of 800/100mg
- allows for convenient, once-daily dosing
- can maintain adequate DRV plasma concentrations for prolonged periods of time
- is well tolerated.
These findings are consistent with recent clinical data on once-daily use of DRV/r 800/100mg in treatment-naive patients participating in the Phase III ARTEMIS trial.4
With its long half-life and suitable exposure following once-daily dosing, DRV is
expected to provide benefits in HIV-1-infected patients, and may support the
long-term sustainability of combination ARV therapy.
Introduction
· Once-daily protease inhibitor (PI) dosing offers the advantages of a reduced pill burden, a less complicated treatment regimen and improved convenience (which may improve adherence), compared with twice-daily PI dosing. In addition, the daily dose of ritonavir needed may be halved for some PIs.
· DRV/r at a dose of 600/100mg bid has been approved in the USA,1 Europe2 and other countries for the treatment of HIV-1 infection in treatment-experienced adults.
· Coadministration of DRV with ritonavir decreases DRV clearance, increasing the DRV terminal elimination half-life (t1/2term) to approximately 15 hours.3
· Once-daily DRV/r may thus present an appropriate therapeutic strategy. DRV/r 800/100mg qd has been evaluated in patients with a broad range of treatment experience, including
- treatment-naive patients in the Phase III ARTEMIS trial4
- antiretroviral (ARV)-experienced patients in the POWER 1 and 2 trials.5,6
· In this study (TMC114-C137), we compared the steady-state plasma pharmacokinetics of DRV for various doses of DRV/r given once or twice daily in
healthy volunteers.
Methods
TMC114-C137 was a Phase I, open-label, parallel-group, dose-ranging study with the primary objective of determining the plasma pharmacokinetics of various oral DRV/r treatment regimens.
Secondary objectives were to assess
- dose-proportionality of DRV exposure for the investigated treatment regimens
- percentage of unchanged DRV excreted in urine
- safety and tolerability of the investigated treatment regimens.
The study population consisted of 40 HIV-negative volunteers grouped into five
panels, each composed of eight healthy adults. During five parallel sessions, volunteers were treated from Day 1 to 7 with the following DRV/r treatment regimens: A) 400/100mg qd; B) 800/100mg qd; C) 1200/100mg qd; D) 400/100mg bid and E) 800/100mg bid (Figure 1). Administration of ritonavir continued until Day 11 of each session.
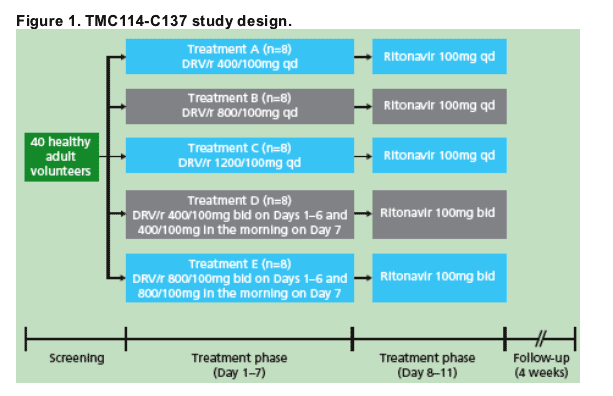
On Days 1 and 7 of each treatment, DRV and ritonavir were coadministered after a standard breakfast.
On Day 7 of each session, the complete urinary output during the intervals 0-12 hours and 12-24 hours (for Treatments A, B and C) or during the interval 0-12 hours (for Treatments D and E) after the morning intake of Day 7 was collected.
DRV and ritonavir plasma concentrations and DRV urine concentrations were determined using validated liquid chromatography tandem mass spectrometry methods
- lower limit of quantification in plasma was 10ng/mL for DRV and 5ng/mL for ritonavir
- lower limit of quantification in urine was 20ng/mL for DRV.
Steady-state plasma PK profiles of DRV were determined up to 120 hours postdose on Day 7 to evaluate the time taken for DRV concentrations to fall below the prespecified threshold of 55ng/mL, based on the protein-binding corrected in-vitro EC50 value for wild-type HIV-1 protease.7
PK analyses were performed as follows
- WinNonlin ProfessionalTM (version 3.3; Pharsight Corporation, Mountain View,
California, USA) software was used to compare Treatment A versus B, A versus C, B versus C and D versus E for DRV
- primary PK parameters included the predose plasma concentration (C0h), dose-normalized minimum plasma concentration between 0 hour and the dosing interval (Cmin), the maximum plasma concentration (Cmax) and the area under the plasma concentration-time curve on the logarithmic scale calculated up to 12 hours postdose (AUC12h) for twice-daily regimens and up to 24-hours postdose (AUC24h) for once-daily regimens
- time to Cmax and t1/2term were also assessed, as well as urine parameters on Day 7, including the percentage of the dose excreted in the urine 0-12 hours postdose (Durine,0-12h), Durine,12-24h (for once-daily dosing) and Durine,total
- the least square means (LSM) of the primary parameters for each treatment group was estimated with a linear, mixed-effects model, controlling for dose as a fixed effect
- a 90% confidence interval (CI) was constructed around the difference between the LSM of test and reference, and both the difference between the LSM and the 90% CI limits were retransformed to the original scale.
Safety and tolerability evaluations were recorded throughout the study, including a 4-week follow-up period after ritonavir administration.
RESULTS
Study population
A total of 40 volunteers, 34 (85%) of whom were male, participated in the trial; 39 were Caucasian and one was African-American. The median ages in the five panels ranged from 35 to 44 years.
All volunteers had negative HIV serology, negative hepatitis B surface antigen and negative hepatitis B core antibody IgM.
Data were excluded from the PK descriptive statistics and statistical analyses for
- one volunteer in the Treatment B group who received disallowed comedications for this study
- one volunteer in the Treatment C group who tested positive for hepatitis C antibody.
DRV PK parameters
On Day 7, Cmax values for DRV were reached between 2.5 and 4 hours postdose for all treatments.
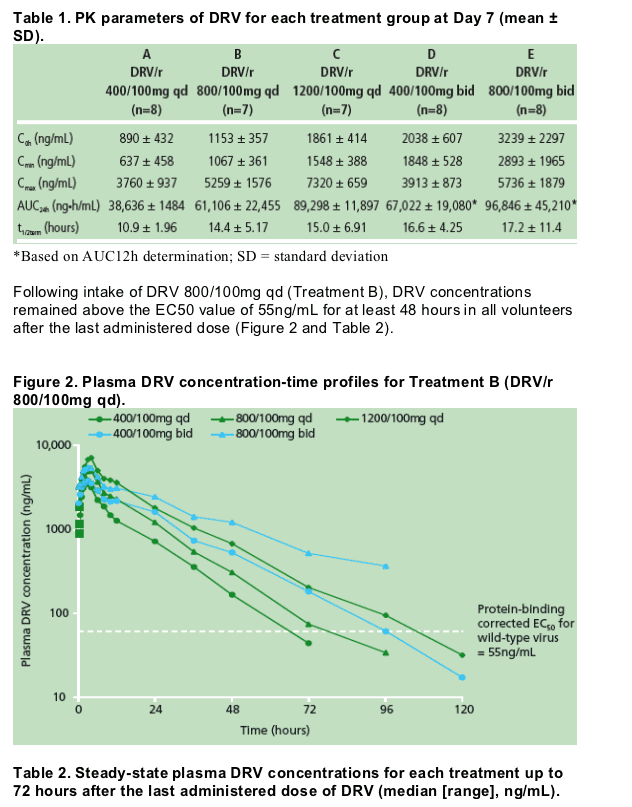
Table 1 shows the PK parameters for DRV at Day 7
- mean concentrations of DRV after Treatment B and Treatment C were comparable
- mean C0h for all treatments exceeded the protein-binding corrected in-vitro EC50 value of 55ng/mL.7
Following intake of DRV 800/100mg qd (Treatment B), DRV concentrations remained above the EC50 value of 55ng/mL for at least 48 hours in all volunteers after the last administered dose (Figure 2 and Table 2).
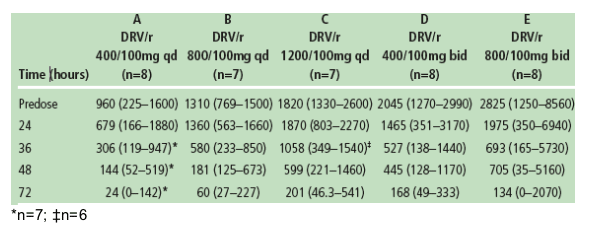
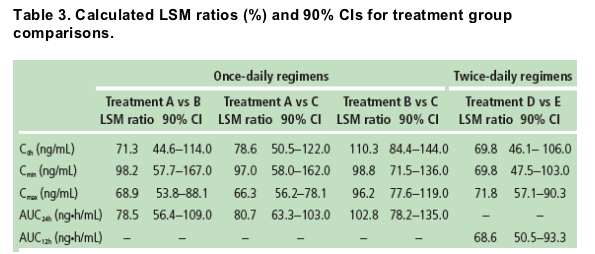
Based on the ratio of LSM and associated 90% CIs of the dose-normalized parameters at Day 7 (Table 3)
- AUC24h and Cmin of DRV increased in proportion with dose for the once-daily regimens
- a less than dose-proportional increase in Cmax was noted for the once-daily regimens (between Treatment A and B, and between Treatment A and C)
- a dose-proportional increase in Cmin and less than dose-proportional increases in Cmax and AUC12h were observed for the twice-daily regimens.
The mean t1/2term of DRV ranged from 10.9 to 17.2 hours. No influence of ritonavir dose regimens on DRV t1/2term was observed.
The mean urinary excretion of DRV was similar for all treatments and was 3-5% of the dose administered within a dosing interval (Table 4).
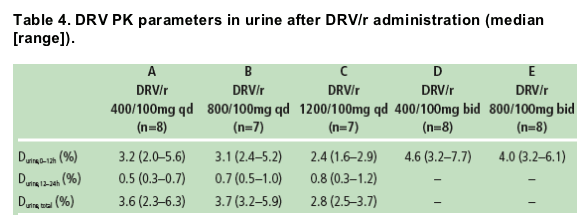
Ritonavir PK parameters
On Day 7, Cmax values for ritonavir were reached between 4 and 6 hours postdose for all treatments.
Daily ritonavir exposure for the twice-daily regimens was approximately twice that observed for the once-daily regimens (Table 5). At Day 7, mean AUC24h for ritonavir was comparable for the once-daily regimens.
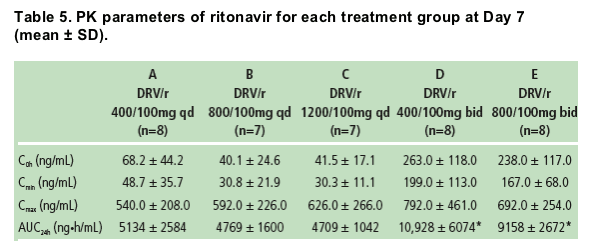
Safety and tolerability
Overall, 27 of the 40 volunteers (67.5%) reported at least one adverse event (AE) during the study. During the treatment phase of the study
- the majority of AEs (31/34 [91%]) were grade 1 or 2 in severity
- there was no dose-response relationship for the incidence of AEs or laboratory abnormalities with the DRV/r treatment regimens.
All changes in laboratory evaluations were grade 1 and grade 2, with the exception of
- treatment-emergent grade 2 and 3 increases in cholesterol reported during DRV/r administration in 7/40 (18%) and 1/40 (3%) volunteers, respectively
- a grade 3 increase in lipase levels at Day 7 following completion of DRV/r administration, considered possibly related to study drug, in one volunteer in the Treatment A group; the volunteer had grade 1 lipase levels at screening and discontinued on Day 12.
No serious AEs or grade 4 laboratory abnormalities in any parameter were observed during the study.
REFERENCES
1. Tibotec Inc. PREZISTA (darunavir) Prescribing Information. February 2008 [accessed 10 March 2008]. Available from: http://www.prezista.com/prezista/.
2. PREZISTA (darunavir) Summary of Product Characteristics. February 2007 [accessed 10 March 2008].
Available from: http://www.emea.europa.eu/humandocs/PDFs/EPAR/prezista/H-707-PI-en.pdf.
3. Hoetelmans R, et al. 10th Conference on Retroviruses and Opportunistic Infections, Boston, USA, 10-14 February 2003. Abstract 549.
4. DeJesus E, et al. 47th Interscience Conference on Antimicrobial Agents and Chemotherapy, Chicago, IL, USA,
17-20 September 2007. Abstract H-718b.
5. Katlama C, et al. AIDS 2007;21:395-02.
6. Haubrich R, et al. AIDS 2007;21:F11-F18.
7. Sekar V, et al. 15th Conference on Retroviruses and Opportunistic Infections, Boston, MA, USA, 3-6 February 2008. Abstract L-103.
|
| |
|
|
|
|
|