| |
The Accelerating Pace of HCV Research: A Summary of the 15th International Symposium on Hepatitis C Virus and Related Viruses
|
| |
| |
Gastroenterology
January 2009
Robert E. Lanford, Matthew J. Evans, Volker Lohmann, Brett Lindenbach_, Michael Gale Jr, Barbara Rehermann#, Kyong-Mi Chang, Stanley M. Lemon
Southwest Foundation for Biomedical Research, San Antonio, Texas
Department of Microbiology, Mount Sinai School of Medicine, New York, New York
Department of Molecular Virology, University of Heidelberg, Heidelberg, Germany
_ Section of Microbial Pathogenesis, Yale University School of Medicine, New Haven, Connecticut
Department of Immunology, University of Washington, Seattle, Washington
Immunology Section, Liver Diseases Branch, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Department of Health and Human Services, Bethesda, Maryland
University of Pennsylvania and Philadelphia VA Medical Center, Philadelphia, Pennsylvania
Institute for Human Infections & Immunity, University of Texas Medical Branch, Galveston, Texas
Article Outline
♦ Early Steps in Infection: Viral Entry
♦ Viral Translation and Protein Processing
♦ RNA Replication
♦ Virus Structure, Assembly, and Release
♦ Innate Immune Responses
♦ Adaptive Immune Responses
♦ Virus-Host Interactions and Pathogenesis
♦ Carcinogenesis
♦ Advances in Antiviral Therapies
♦ Vaccine Development
♦ Summary and Future Directions
Almost 800 research scientists convened in San Antonio in early October 2008 to share recent results and new insights concerning hepatitis C virus (HCV). The 15th International Symposium on Hepatitis C Virus and Related Viruses opened with a mini-symposium recognizing progress made over the past 20 years since the discovery of HCV. This was followed by 10 keynote talks, 61 oral presentations, and 379 poster presentations over the succeeding 4 days. Our intent is to succinctly summarize the most notable findings reported, a task made extraordinarily difficult by the volume, diversity, and quality of science presented. At best, this cursory overview vividly demonstrates the robust pace of the research that is unraveling the mysteries of this unique and potentially deadly human pathogen.
Early Steps in Infection: Viral Entry
The opening scientific session focused on host factors involved in HCV cell entry (Figure 1). Jane McKeating (University of Birmingham, Birmingham, England) began the session with an overview of current understanding of the entry process that highlighted several recent advances from her laboratory. These included the development of novel anti-CD81 antibodies that specifically detect distinctly localized, higher-order, complexed forms of CD81. These antibodies blocked viral entry with different kinetics in synchronized infections, suggesting that CD81 is required at multiple stages of the entry process or that distinct domains selectively blocked by these antibodies are required for specific steps of the infection process. McKeating also described studies of direct HCV cell-cell spread that showed that this process requires intact adherens junctions, a low pH step, and very-low-density lipoprotein pathways; these observations elucidate this poorly defined mode of HCV spread.
Figure 1. Overview of HCV cell entry. The HCV entry process appears to require numerous interactions with host factors, both soluble and on the cell surface. The incoming virion, which appears to associate with apolipoprotein complexes, may first bind a host cell by interacting with the low-density lipoprotein receptor (LDL-R) and glycosaminoglycans (GAGs) on the cell surface. Many HCV entry factors are required after virion binding to the host cell. These include an early requirement for CD81 and scavenger receptor class B type 1 (SR-B1), followed by a later utilization of the tight junction protein CLDN1. It is yet to be determined when the latest HCV entry factor OCLN, also a tight junction protein, is required. Such interactions result in endocytosis of the virion, where it fuses with an endosomal membrane upon acidification to release the viral nucleocapsid.
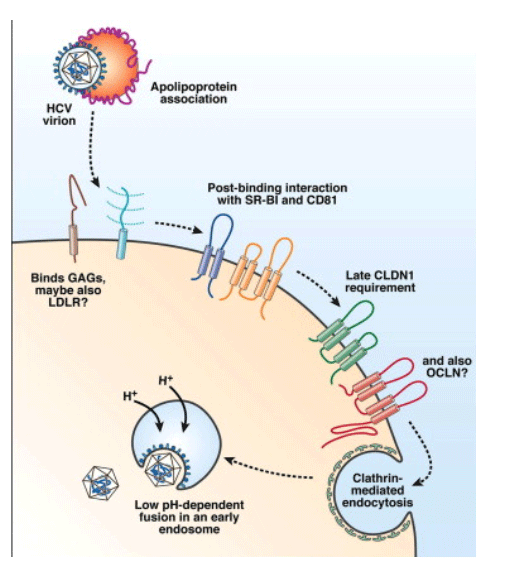
Philip Meuleman (Ghent University, Ghent, Belgium) presented a study in which HCV infection of mice with humanized livers was blocked in a dose-dependent manner by prior injection of anti-CD81 monoclonal antibodies. Not only was this the first in vivo evidence that CD81 is an essential HCV entry factor, but it also provided the first proof that entry inhibitors can be effective in blocking infection in animals. However, anti-CD81 antibodies failed to inhibit HCV infection when administered after virus challenge, so anti-CD81 antibodies might only be useful in specific clinical settings, such as preventing graft infection following transplantation.
Julia Bitzegeio (Twincore Center, Hannover, Germany) described the identification of mutations in the E1 and E2 envelope proteins that, when combined, increased the ability of HCV to infect cells that express murine CD81. This expansion of HCV tropism was not associated with reductions in the ability of the mutant virus to use human CD81 for cell entry. The mutations appeared to act by increasing the affinity of the envelope proteins for CD81, as the mutant viruses were less susceptible to inhibition by anti-CD81 antibodies.
Finally, a poster by Tianyi Wang and colleagues (University of Pittsburgh, Pittsburgh, PA) described the requirements for various tight junction proteins in HCV entry. By silencing such factors in Huh-7 cells, they identified a requirement not only for claudin-1 (CLDN1), but also occludin (OCLN), in entry of cell culture-infectious virus (HCVcc) and pseudotyped viral particles (HCVpp). This finding was similar to that described by Charles Rice (Rockefeller University, New York, NY) in his plenary lecture in the opening mini-symposium, in which he presented new data showing that OCLN is an essential cell entry factor in human cells. Rice also reported that expression of human OCLN, in combination with CD81, SR-B1, and CLDN1, rendered murine cells permissive for HCVpp entry. Furthermore, although human CD81 and OCLN were required for full permissivity, the murine and human versions of CLDN1 and SR-BI allowed equal amounts of HCVpp entry, suggesting that CD81 and OCLN are the most important determinants of species-specific viral entry.
Viral Translation and Protein Processing
Darius Moradpour (University of Lausanne, Lausanne, Switzerland) began the session with an overview of HCV protein membrane topology, describing what is known of the mechanisms that determine these associations. He included a description of recent collaborative work with Francois Penin (University of Lyon, Lyon, France) on the membrane association mechanisms of the NS3-4A protease complex. This is mediated by 2 structural determinants: an amphipathic a-helix within the N-terminal part of NS3 and the N-terminal transmembrane a-helix of NS4A. These structures function in unison to position the NS3-4A active site in close proximity to the membrane, which may have consequences related to the timing of downstream trans-cleavages, and also confer additional selectivity to the protease by restricting its access to cellular proteins. Moradpour proposed a dynamic model for the membrane association, processing, and structural organization of NS3-4A with important implications for the functional architecture of the HCV replication complex, proteolytic targeting of host factors, and potential antiviral drug design.
Norbert Tautz (University of Lubeck, Lubeck, Germany) presented a genetic analysis of the NS2-3 protease. By screening a panel of deletion mutants, Tautz and colleagues found that NS2 protease activity requires only the first 2 N-terminal residues of NS3, indicating that the downstream N-terminal NS3 domain functions only as a cofactor that promotes efficient cleavage at the NS2-3 junction. Consistent with this, he reported that an NS3 domain containing the Zn-binding site stimulated NS2 protease activity in trans. These findings suggest that the NS2-3 protease is an attractive antiviral target and will accelerate the development of appropriate inhibitor screening assays. One interesting aspect of Tautz's findings was that the N-terminal NS3 domain has 2 different folds: the first acts directly after translation as the NS2 cofactor, while the alternative fold associates with NS4A to promote full serine protease activity. Subsequent polyprotein processing events are exquisitely regulated in ways that are likely to contribute to overall viral fitness, as indicated by Annette Martin's group's (Institut Pasteur, Paris, France) comparison of NS3/4A-directed cleavages that occur within the polyproteins of HCV and GBV-B (a closely related hepatotropic flavivirus).
RNA Replication
The session on HCV RNA replication was not dominated by a major breakthrough, but a series of reports improved our understanding of the functions of nonstructural proteins and associated host factors in viral replication. Several presentations focused on NS4B, which induces vesicular membrane rearrangements (the so-called "membranous web") via an unknown mechanism. Daniel Jones (MRC Virology Unit, Glasgow, Scotland) reported that some deleterious mutations in the C-terminus of NS4B can be rescued by trans-complementation with wild-type NS4B. This advance allows a more detailed, mechanistic analysis of the functions of this protein. Interestingly, one mutant gave rise to significantly increased viral yields, arguing for a role of NS4B beyond RNA replication. This is consistent with previous studies showing important roles for other nonstructural proteins (NS2, NS3, and NS5A) in virion assembly. Posters from Jerome Gouttenoire (University of Lausanne, Lausanne, Switzerland) and Hans C. Van Leeuwen (Leiden University, Leiden, The Netherlands) suggested that the C-terminus of NS4B forms an amphipathic a-helix that associates tightly with membranes, whereas Kouacou Konan (Pennsylvania State University, University Park, PA) presented data suggesting that this NS4B domain is critically involved in the formation of replication complexes. Several reports from Jeffrey Glenn's group (Stanford University, Stanford, CA) suggested that NS4B represents a viable target for antiviral drug development. His group identified mutations within NS4B that confer resistance to a particular compound and also screened compounds for their ability to inhibit the recently identified RNA-binding activity of NS4B.
Despite these advances, a major bottleneck in HCV research remains: the limited availability of viral isolates that are capable of undergoing the entire viral life cycle in cell culture. Only RNAs derived from molecular clones that contain sequences that encode the nonstructural proteins of the JFH1 virus, a genotype 2a virus identified in a Japanese patient with fulminant hepatitis, are capable of highly efficient virus production (>104 infectious virus particles/mL) when cultured cells are transfected. Previous studies have shown that an important determinant of this unique property of JFH1 resides in the sequence encoding the viral RNA-dependent RNA polymerase NS5B. Melanie Schmitt (University of Heidelberg, Heidelberg, Germany) described a functional and structural analysis of the JFH1 polymerase. She suggested that this enzyme is particularly efficient in initiating RNA synthesis de novo and that this might contribute to the unique replication phenotype of JFH1 virus in cell culture. The JFH1 polymerase adopts the most closed conformation of all available HCV RNA-dependent RNA polymerase structures, potentially enhancing its ability to bind substrates for de novo initiation. These are important studies, because a better understanding of the structural features of the JFH1 polymerase might indicate ways to alter the polymerases of other HCV strains to make them replicate better in cultured cells. A poster presented by Nobuyuki Kato and colleagues (Okayama University, Okayama, Japan) reported the molecular cloning of a novel genotype 1b isolate that replicates in cultured cells without a requirement for adaptive mutations and appears to produce infectious virus with low efficiency.
This session also focused on the mechanisms of action of cyclosporin A and related nonimmunosuppressive derivatives, such as Debio-025 and NIM811, which are being studied as candidate antiviral therapies. Recent studies indicate that cyclophilin A, rather than cyclophilin B, is the key mediator of inhibition of HCV replication by cyclosporin A. Three separate presentations, by Phillip Gallay (Scripps Research Institute, La Jolla, CA), Artur Kaul (University of Heidelberg, Heidelberg, Germany), and Zhe Liu (Florida State University, Tallahassee, FL), indicated that the peptidylprolyl-cis/trans isomerase activity of cyclophilin A is critical for HCV replication, although it is not completely clear which viral protein is modified by the isomerase. Published studies indicating a direct interaction of cyclophilin A with the viral polymerase NS5B were substantiated by data from Liu and colleagues suggesting that cyclophilin A specifically recruits NS5B to the viral replication complex. Still, data from Kaul and from Kai Lin's group (Novartis, Cambridge, MA) indicate that mutations in the viral genome conferring resistance to cyclosporin A or its derivatives map primarily to NS5A and only secondarily to NS5B. Interestingly, Kaul reported that a particular mutation in NS5A that confers resistance to Debio-025 also significantly slows processing at the NS5A-5B junction.
Virus Structure, Assembly, and Release
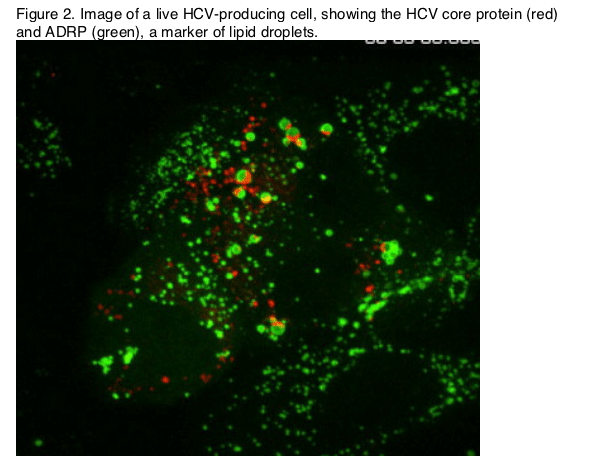
The structure, assembly, and release of HCV particles are perhaps the least understood aspects of the virus life cycle. However, the recent development of fully infectious virus culture systems has provided the tools necessary to study these processes, and exciting progress was reported at this symposium. Richard Kuhn (Purdue University, West Lafayette, IN) began this session with a review of the structural basis for dengue virus maturation, which involves significant rearrangements of the envelope glycoproteins. Whether this is a general feature of all flaviviruses, including HCV, remains to be determined, but the presentation stimulated numerous hallway discussions. Brett Lindenbach (Yale University, New Haven, CT) subsequently reported the ability to image live cells infected with HCV that contains a functional, tetracysteine-tagged core protein (Figure 2). Notably, the core protein localized to stationary lipid droplets and bright punctae that trafficked via microtubules. This system shows promise for the study of the dynamics of core protein in live virus-producing cells.
The presentations that followed were a "coming-out party" for the nonstructural proteins that have a role in virus particle production; the belle of this year's ball was NS2. In oral and poster presentations, several groups described NS2 mutations that severely compromised virus production. MinKyung Yi (University of Texas Medical Branch, Galveston, TX) and Vlastimil Jirasko (University of Heidelberg, Heidelberg, Germany) showed that NS2 defects could be complemented in trans. Yi also reported that an NS2-mediated defect in virus production could be suppressed by second-site mutations in NS2 and NS5A. Lindenbach's group reported several additional NS2 mutants with defects in virus assembly and identified second-site suppressor mutations in E1, E2, NS3, and NS4A. Thus, NS2 likely interacts with multiple structural and nonstructural proteins during infectious particle production. Of particular interest were reports of an interaction between p7 and NS2. Philip Tedbury (University of Leeds, Leeds, England) and Costin Popescu (Institut de Biologie de Lille, Lille, France) provided genetic and cell biological evidence for a functional interaction between the transmembrane domains of NS2 and p7. In addition, Yi and Corine St. Gelais (University of Leeds, Leeds, England) independently showed that the p7 and NS2 mutants continue to produce intracellular, core protein-containing particles. Thus, p7 and NS2 might coordinately contribute to a late step in particle assembly or maturation.
The HCV NS5A protein continues to reveal new secrets about the viral life cycle, and several groups reported genetic evidence that NS5A domain III is an important determinant for virus particle production. Tim Tellinghuisen (Scripps Research Institute Florida, Jupiter, FL) described numerous NS5A mutants with defects in virus assembly and showed that one mutant phenotype could be complemented by replication-defective viral genomes in trans. This result elegantly shows that the role of NS5A in viral genome replication can be uncoupled from its role in particle assembly. Based on this system, Tellinghuisen's group identified several major structural features of NS5A that are independently required for genome replication and virus particle production.
However, according to McKeating, we still do not understand what constitutes an infectious HCV particle. One investigational approach has been to purify and characterize proteins associated with virus particles. The challenges to and progress made with this approach were highlighted in several talks. Somewhat surprisingly, Jieyun Jiang (University of Kentucky, Lexington, KY) and Soren Nielsen (Newcastle University, Newcastle upon Tyne, England) independently reported that viral nonstructural proteins were copurified with virus particles obtained from cell culture or patient-derived material. This is an intriguing finding that highlights the need to further study highly purified virus preparations. Although a growing body of evidence also indicates that HCV particles associate with serum lipoproteins, the nature of these interactions remains unclear. Wagane Benga (Universite Louis Pasteur, Strasbourg, Alsace, France), Vinca Icard (Universite de Lyon, Lyon, France), and Jiang each presented evidence that HCV glycoprotein secretion and virus particle assembly require an intact very-low-density lipoprotein assembly pathway. Thus, HCV appears to represent a new paradigm in virus assembly and host-pathogen interaction. Other host cell proteins also appear to have key roles in assembly and release, as reported by Perdita Backes (University of Heidelberg, Heidelberg, Germany), who showed that HCV assembly depends on annexin A2, a host protein that colocalizes to viral replication complexes and appears to be recruited by NS5A.
Finally, Jens Bukh and his colleagues (Copenhagen University Hospital, Copenhagen, Denmark) reported the development of recombinant, chimeric, full-length genomes encoding structural proteins from genotypes 1-7 HCV and nonstructural proteins from JFH1 virus. These chimeric genomes produce infectious virus in cell culture and should prove to be valuable tools for future research.
Innate Immune Responses
This session featured new information on mechanisms by which innate immune responses are triggered and controlled by HCV. A keynote presentation by Glenn Randall (University of Chicago, Chicago, IL) summarized the current understanding of the mechanisms by which interferon (IFN) controls HCV infection and described how 2 key IFN-stimulated genes (ISGs), ISG15 and USP18, function within a protein modification cycle to regulate hepatic innate immunity and virus infection. A notable aspect of the ISG15/USP18 program is that each protein possesses enzymatic activities that could be targeted therapeutically in an effort to enhance innate immunity and thus limit HCV infection.
HCV triggers the innate immune system, in part, through the cellular RIG-I helicase, which recognizes and binds to HCV RNA and signals the downstream expression of IFN genes and ISGs. How does RIG-I recognize HCV RNA? Takeshi Saito (University of Washington, Seattle, WA) presented new findings that identify the poly-U/UC motif within the 3' nontranslated segment of the HCV genome as a pathogen-associated molecular pattern that is recognized and bound by RIG-I in the context of a 5' triphosphate, thereby triggering IFN expression. Similar poly-U motifs found in the genome of other RNA viruses also stimulate RIG-I signaling. Thus, poly-U motifs within RNAs containing 5' triphosphates are recognized as "non-self" pathogen-associated molecular patterns by RIG-I. HCV can block the actions of RIG-I signaling by targeting and cleaving the RIG-I adaptor protein known as MAVS, IPS-1, or Cardif through the actions of the viral NS3/4A protease. Stacy Horner (University of Washington, Seattle, WA) described how NS3/4A targets this protein, which is located on the mitochondria membrane. Both mitochondrial localization and MAVS/IPS-1/Cardif cleavage were shown to be dependent on the a0 helical domain located within the N-terminus of NS3. The presence of a mitochondrial localization domain within the NS3/4A protease is thus an important factor in the disruption of RIG-I signaling by HCV. This feature of NS3/4A is reminiscent of a mitochondrial localization domain reported previously within the hepatitis A virus 3ABC protease, which also targets MAVS/IPS-1/Cardif for cleavage.
In addition to targeting MAVS/IPS-1/Cardif, studies with HCV replicons suggest that NS3/4A cleaves the cellular Toll-like receptor (TLR)-3 adaptor protein TRIF. Thus, it might also target additional cellular proteins for proteolysis. Erwin Benndorfer (Heinrich-Heine University, Dusseldorf, Germany) reported that the NS3/4A protease induces epidermal growth factor signaling and cell proliferation by reducing the abundance of the TC-PTP protein phosphatase, a key regulator of epidermal growth factor signaling. The expression of NS3/4A alone or in the context of HCV RNA replication resulted in reduced TC-PTP expression and increased phosphorylation of its protein targets in cultured cells and in vivo. Although these observations are consistent with potential NS3/4A proteolysis of TC-PTP, it is not clear whether NS3/4A actually cleaves TC-PTP or indirectly regulates the stability of the protein. NS3/4A regulation of TC-PTP could influence HCV replication and contribute to pathogenesis by disrupting cellular homeostasis and growth control.
Ribavirin significantly increases the therapeutic actions of IFN in HCV-infected patients by an unclear mechanism. Emmanuel Thomas (National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD) described a novel property of ribavirin, showing that it induces the expression of a subset of ISGs independently of IFN or signaling by either TLRs or RIG-I. ISG expression was transient following treatment of cultured cells with ribavirin, consistent with subsequent induction of a negative regulator of ISG expression. The ability of ribavirin to stimulate ISG expression may explain how it improves the therapeutic efficacy of IFN. However, further studies are needed to determine how it activates ISG expression and whether this enhances innate immune control of HCV replication.
The innate immune response has been studied mostly in the setting of acute infection, and its role in chronic infection is not well understood. Golo Ahlenstiel (National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD) assessed natural killer cell function in patients with chronic HCV infection. Natural killer cells from these patients were activated and exhibited increased cytotoxicity, but not increased IFN-γ expression. This "polarized" phenotype correlated with the degree of liver injury, suggesting that natural killer cells contribute to liver inflammation in chronic HCV infection.
Adaptive Immune Responses
The session opened with a keynote presentation by Patrick Bertolino (University of Sydney, Sydney, Australia), who reviewed unique aspects of the intrahepatic immune system. He presented data showing that both primary activation of CD8+ T cells and removal of activated CD8+ T cells take place within the liver. These properties of the liver likely contribute to its role in inducing tolerance. Several of the subsequent presentations focused on viral mutations that may promote escape from adaptive immune responses. Thomas Kuntzen (Harvard University, Cambridge, MA) reported findings from large-scale, full-length HCV sequencing studies that revealed distinct patterns of HCV sequence evolution in the context of the HLA haplotype of the infected host; these provide indirect evidence of selection pressure by HLA-restricted CD8+ T cells. HLA-B27 stood out as one of the alleles most strongly associated with viral clearance. Its apparent protective role is consistent with the observation that viral escape from HLA-B27-restricted, NS5B-specific CD8+ T-cell responses is difficult because of the high cost to viral fitness and requirement of several clustered mutations, as reported by Eva Dazert (University of Heidelberg, Heidelberg, Germany).
In contrast to CD8+ T cells, Michael Fuller (Nationwide Children's Hospital, Columbus, OH) reported that CD4+ T cells mediate less selection pressure, based on a study of HCV-infected chimpanzees. Moreover, CD4+ T cells can be compartmentalized to the liver and remain entirely undetectable in the blood during the first year of infection. Eva Billerbeck (University of Freiburg, Freiburg, Germany) suggested that hepatic compartmentalization was especially prominent for CD4+ T cells that produce the proinflammatory cytokine interleukin-17, whereas Yu-Hoi Kang and colleagues (University of Oxford, Oxford, England) described CD8+ T cells with a liver-homing, CD161/CXCR6 phenotype that are deficient in cytotoxic effector functions. Nobuhiro Nakamoto (University of Pennsylvania, Philadelphia, PA) also showed that intrahepatic CD8+ T cells become functionally impaired with up-regulation of programmed death-1 (PD-1) and cytotoxic T-lymphocyte antigen 4 expression. Simultaneous blockade of both the PD-1 and the cytotoxic T-lymphocyte antigen 4 pathways was required to fully restore their function in vitro.
Humoral immune responses have been assessed using in vitro assays that evaluate the ability of antibodies to block infection with HCVcc or entry of HCVpp into cultured hepatoma cells. Mirjam Zeisel (Universite Louis Pasteur, Strasbourg, Alsace, France) showed that neutralizing antibodies disrupt viral entry steps that require interactions between HCV and CD81 and between SRB1 and CLDN-1, as well as steps that occur after binding and during membrane fusion. Consistent with the paradigm of viral escape, Zhenyong Keck (Stanford University, Stanford, CA) reported that mutations in HCV E2 that lie outside the CD81 binding site prevent recognition by neutralizing antibodies and are tolerated by the virus, despite structural changes and compromised fitness due to reduced binding to CD81.
Virus-Host Interactions and Pathogenesis
The session opened with a keynote lecture by Kunitada Shimotohno (Keio University, Tokyo, Japan) describing the role of lipid droplets in infectious virion production. Similar to the viral assembly session, several of the subsequent presentations focused on the role of cellular lipid metabolism/trafficking in different steps of the HCV lifecycle. Yutaka Amako (University of California at San Diego, San Diego, CA) analyzed viral ribonucleoprotein complexes by tandem mass spectrometry and identified oxysterol-binding protein as a host factor required for viral replication and virion maturation. Eva Herker (Gladstone Institute, San Diego, CA) reported that the assembly of infectious HCV depends on active diacyl glycerol acyltransferase (DGAT1)-mediated lipid droplet formation, induced by the viral core protein. On the other hand, carboxyesterase 1, a key enzyme in lipid and cholesterol storage, was reported by John Pezacki (University of Ottawa, Ottawa, Ontario, Canada) to block formation of the HCV replication complex. This enzyme was identified by activity-based profiling of serine hydrolases and shown to occupy the ER-derived membrane surrounding lipid droplets where the HCV core is known to localize. Efficient replication of HCV in cell culture was also reported by Laurent Chatel-Chaix (University of Montreal, Montreal, Quebec, Canada) to be dependent on Y-box binding protein-1 (YB-1), which was found to associate with NS3/4A. RNA helicase A was also reported to associate with NS3/4A and may promote replication.
Carcinogenesis
A hallmark of this meeting was a session on viral carcinogenesis, a much-neglected area of HCV biology despite the growing clinical burden of HCV-associated hepatocellular carcinoma (HCC) worldwide. In a keynote talk, Mark Harris (University of Leeds, Leeds, England) reviewed the various cellular pathways implicated in HCV-associated HCC and the viral proteins involved in dysregulation of these pathways, which include p53 (core, NS3, NS5A), transforming growth factor β (core, NS5A), retinoblastoma tumor suppressor protein (Rb) (NS5B), epidermal growth factor (NS5A), and β-catenin (NS5A). Despite these disruptions, HCC does not develop in all HCV-infected patients, suggesting that additional factors are involved. Remarkably, Robert Lanford (Southwest Foundation for Biomedical Research, San Antonio, TX) showed that liver cancer can develop in chimpanzees many years after experimental infection with HCV (1/47) or hepatitis B virus (1/8), even in the absence of cirrhosis.
Multiple reports have suggested that specific host-pathogen interactions contribute to carcinogenesis. Urszula Hibner (University of Montpellier, Montpellier, France) reported that HCV activates calpain, a cellular protease that cleaves and activates Bid (a proapoptotic molecule in the Bcl2 family), thereby promoting resistance to Fas-mediated apoptosis. Francois Duong (University Hospital Basel, Basel, Switzerland) reported that HCV up-regulates protein phosphatase 2A, thereby reducing epigenetic histone modification and H2AX phosphorylation and impairing DNA repair. Men are at increased risk for HCC; Ratna Ray (Saint Louis University, St Louis, MO) showed that expression of the HCV core protein increases androgen receptor signaling to increase vascular endothelial growth factor expression and angiogenesis and promote HCC progression.
Several other reports specifically implicated NS5A in the development of HCV-related HCC. Jameel Mankouri (University of Leeds, Leeds, England) reported that NS5A suppresses voltage-gated potassium channels to protect cells against apoptosis via p38 mitogen-activated protein kinase membrane insertion; this effect could contribute to viral persistence as well as cellular transformation. NS5A also dysregulates the transcriptional activity of RNA polymerase I, according to Asim Dasgupta (University of California at Los Angeles, Los Angeles, CA). RNA polymerase I catalyzes ribosomal RNA transcription and ribosome biogenesis; changes in its transcription rate could alter the growth rate of infected cells. Interestingly, Herve Lerat (Hopital Henri Mondor, Creteil, France) showed that the transcriptional transactivating property of NS5A was increased in variants isolated from tumor tissue, compared with adjacent nontumor tissue. Finally, Keigo Machida (University of Southern California, Los Angeles, CA) suggested that alcohol and NS5A act synergistically to produce cancer; alcohol promoted hepatocyte transformation in NS5A transgenic mice by inducing TLR4 signaling and up-regulating expression of Nanog, a novel TLR4 downstream gene encoding a stem cell marker.
Advances in Antiviral Therapies
The strong focus on development of HCV antiviral agents was vividly shown by the 77 oral and poster presentations on this topic. The session opened with the Giovanni Migliaccio Memorial Lecture, delivered by Daniel Lamarre (University of Montreal, Montreal, Quebec, Canada). Lamarre emphasized the difficulties in bringing candidate therapeutics from discovery through clinical trials. Whereas 24 antiviral agents have been approved for treatment of patients with human immunodeficiency virus in the past 25 years, only one anti-HCV compound has advanced to phase 3 trials in the 20 years since the discovery of HCV. However, the field is rapidly accelerating, as more than 20 anti-HCV agents are in various stages of clinical trials.
In the area of protease inhibitors, Steve Ludmerer (Merck Research Laboratories, Rahway, NJ) presented in vivo data on the antiviral activity of the macrocyclic inhibitor MK-7009. Low nanomolar concentrations of MK-7009 have activity in the replicon model. In HCV-infected chimpanzees, viremia was suppressed to levels below the limits of detection (>5 log10 decline) within 5 days. Although a resistant variant, NS3-R155K, was present at detectable levels before dosing and rapidly emerged during testing, the inhibitor prevented a rebound in viremia during the dosing phase. Following treatment, the viral population rapidly shifted back to wild type, although resistant virus remained detectable for months. Origene Nyanguile (Tibotec, Mechelen, Belgium) presented studies on the structure-activity relationship of a potent non-nucleoside polymerase inhibitor in the 1,5-benzodiazepine class. This dioxythiopyrane-fused benzodiazepine analogue is a potent inhibitor of the genotype 1b replicon, with a 50% effective concentration of 20 nmol/L. However, it has significantly less activity against genotype 1a viruses. The inhibitor binds to the palm domain of NS5B, which is adjacent to the active site (non-nucleoside polymerase inhibitor 3 site). Genotype 1a and 1b viruses differ by only a few residues in this region, but a structure-function analysis did not completely elucidate the basis of the differential genotype sensitivity.
Safety, Tolerability and Antiviral Activity of MK-7009, a Novel ...
Nov 1, 2008 ... MK-7009 is generally well tolerated at all doses studied -- No dose dependent safety ... A Phase IIa study of MK-7009 administered with ...
www.natap.org/2008/AASLD/AASLDa09.htm
Safety, Tolerability, and Pharmacokinetic Data Following Single- and Multiple-Dose Administration of MK-7009, a Hepatitis C Virus Non-structural 3/4a Protease Inhibitor, to Healthy Male Subjects (1910) - (11/14/08)
Two presentations described new replication inhibitors that appear to target NS5A. Although NS5A function is still poorly understood, increasing evidence implicates NS5A in multiple essential viral functions, including modifying the IFN response, facilitating RNA replication, and assembling the virus. Arrow Therapeutics (London, England) previously described a first-in-class NS5A inhibitor that has entered clinical trials, and this year Pilar Najarro (Arrow Therapeutics) presented data on a new class of NS5A inhibitor, AZ12781194. The compound exhibits significant genotype specificity with greater potency against genotype 1b than genotype 1a HCV (50% inhibitory concentration, 7 nmol/L vs 1 μmol/L). Resistance mutations mapped to domain I at the N-terminus of NS5A (Y93) and the compound inhibited the formation of p58, a hyperphosphorylated form of NS5A. Min Gao (Bristol-Myers Squibb, Wallingford, CT) described a remarkably potent compound, BMS790052, with a 50% effective concentration that is in the picomolar range against all genotypes (50% effective concentration is 9 pmol/L for genotype 1b). Similar to the Arrow compound, resistance mutations mapped to NS5A domain I (L31 and Y94) and inhibited the formation of p58. However, Gao described 2 different compounds that both select for virus containing the Y94H resistance mutation but differentially affect p58 formation, suggesting that antiviral activity might be unrelated to the effect on NS5A hyperphosphorylation. Clinical trial data to be presented at the upcoming American Association for the Study of Liver Diseases meeting will reportedly show a 3.6 log10 decline in viremia following administration of a single dose of this inhibitor.
BMS-790052 is a First-in-class Potent Hepatitis C Virus (HCV) NS5A ...
This new class of drug, the BMS NS5A inhibitor, attracted quite a lot of discussion because of it potent viral load reduction of -3.6 logs, after one dose ...
www.natap.org/2008/AASLD/AASLDa06.htm
New BMS HCV NS5A Inhibitor: From Screen Hit to Clinic
BMS- 790052, is a first-in-class molecule evolved from a screening lead, and is the most potent anti-HCV agent reported to date with an EC50 of 50 pM and 9 ...
www.natap.org/2008/HCV/101408a01.htm
A novel class of viral entry inhibitors based on cyclic D,L-a-peptides was described by Pablo Gastaminza (Scripps Research Institute, La Jolla, CA). The peptides were identified in a screen for inhibitors of JFH1 virus infection; antiviral activity required the cyclic form of the peptide and polymerization into stacked nanotubes. The peptides disrupt an early step in infection, inhibiting HCV-pseudotyped particle entry. Yutaka Takebe (Nippon University, Japan) described a second class of entry inhibitors that are also active against HCV-pseudotyped particles; molecular docking studies implicated a mechanism involving direct CD81 binding.
Steve Polyak (University of Washington, Seattle, WA) described multiple activities (antiviral, immunomodulatory, anti-inflammatory, and antioxidant) of the herbal preparation silymarin. Silymarin is derived from an extract of milk thistle seeds and could contain several active components responsible for its broad range of putatively beneficial effects. Although silymarin had no effect on the replication of full-length HCV replicons, it reduced viral replication in JFH1 virus-infected cells. It also reduced virus-induced oxidative stress, inhibited nuclear factor κB activity, and suppressed cytokine production.
Vaccine Development
In his keynote talk for this session, Rafi Ahmed (Emory University, Atlanta, GA) discussed the features of successful vaccine-induced T-cell-mediated immune responses, using the yellow fever virus vaccine as an example. Unlike HCV, the live attenuated yellow fever virus vaccine induces a robust and durable virus-specific T-cell response within 2 weeks of immunization. The induced T cells display dynamic changes in Ki67, HLA DR, Bcl2, and CD127 expression. Interestingly, vaccine-induced memory CD8+ T cells appeared to be similar to terminally differentiated effectors (CCR7-, CD45RA+), despite efficient functionality. In addition, the coinhibitory receptor PD-1 contributes to reversible T-cell exhaustion. These findings provide a new toolbox to study vaccine response in the future.
The paucity of abstracts submitted for this session shows that HCV vaccine development remains a great challenge; however, several approaches that induce both antibody and T-cell responses were presented. Patricia Vietheer (Burnet Institute, Melbourne, Victoria, Australia) immunized mice with HCV containing deletions in the variable HVR1, HVR2, and igVR domains of the E2 envelope glycoprotein. This resulted in antibodies against conserved regions in E2, overall increased antibody titers in the mice, and greater HCVpp neutralizing activity in vitro. However, concerns for antibody escape were substantiated by Meital Gal-Tanamy (University of Texas Medical Branch, Galveston, TX), who described the in vitro selection of an antibody-resistant mutant following serial passage of virus in the presence of a broadly neutralizing monoclonal antibody, AP33, that recognizes a conserved epitope in E2. Dan Xu (Nationwide Children's Hospital, Columbus, OH) presented the use of recombinant self-complimentary adeno-associated virus (scAAV) as a platform to prime for production of antibodies against linear and conformational HCV E2 epitopes in the guinea pig model.
In the realm of vaccines that induce T-cell responses, Genevieve Inchauspe (Transgene, Strasbourg, Alsace, France) reported the preliminary results of a phase 1 therapeutic vaccine study in which modified vaccinia Ankara encoding HCV proteins (genotype 1b) was administered to treatment-naive patients chronically infected with genotype 1 virus. There were no serious adverse effects, and low-level reductions in viremia were observed (0.6-1.4 log reductions in 6 of 15 subjects). Another clinical study, reported by Heiner Wedemeyer (Hannover Medical School, Hannover, Germany), showed that a peptide vaccine (IC41) did not prevent viremic relapse when combined with standard pegylated interferon/ribavirin therapy.
Finally, adopting a very conventional strategy for vaccine development, Hitoshi Takahashi and his colleagues (Toray Industries, Tokyo, Japan) described their preliminary efforts to produce sufficient HCV particles from JFH1-infected cells for production of an inactivated, cell-culture derived vaccine.
Summary and Future Directions
The meeting concluded with a brief description by Jean-Michel Pawlotsky of plans for next year's HCV meeting, which will be held in Nice, France, on October 3-7, 2009. Given the many advances reported this year, it is likely that the 16th International Symposium in Nice will reveal continued rapid progress in our understanding of HCV cell entry, viral replication mechanisms, and the process of viral assembly and release. Such new knowledge is certain to promote the development of novel HCV treatment strategies, the need for which will continue to increase with the aging of the current cohort of HCV-infected patients.
|
|
| |
| |
|
|
|