| |
The many faces of hepatitis C: Liver disease and type 2 diabetes
|
| |
| |
Editorial
Hepatology Sept 2009
Kerry-Lee Milner 1, Donald Chisholm 1, Jacob George 2
1Garvan Institute of Medical Research, University of New South Wales, Sydney, Australia
2Storr Liver Unit, Westmead Millennium Institute, University of Sydney, Sydney, Australia
Article Text
Epidemiological, clinical, and laboratory data indicate that apart from causing chronic liver injury, hepatitis C virus (HCV) infection is associated with extrahepatic disease. Foremost among the systemic consequences of chronic hepatitis C (CHC) is evidence that infection results in metabolic sequelae with the virus interacting with lipid and glucose metabolism resulting in hepatic steatosis, insulin resistance (IR), type 2 diabetes, and hypocholesterolemia. Multiple cross-sectional and longitudinal[1] studies have identified the association between CHC, IR, and type 2 diabetes. The prevalence of diabetes in CHC is between 19%-33%,[2][3] is specific to hepatitis C compared to other chronic liver diseases such as chronic hepatitis B, is independent of the severity of liver fibrosis, and its incidence is increased in patients with CHC after liver transplantation.[4] A causative association is further suggested by the knowledge that treatment-induced viral eradication results in improvements in insulin sensitivity.[5]
Abbreviations
CHC, chronic hepatitis C; HCV, hepatitis C virus; HOMA-IR, homeostasis model of insulin resistance; HOMA-B, homeostasis model of cell function; IR, insulin resistance; IL, interleukin; TNF-, tumor necrosis factor-alpha.
In type 2 diabetes not associated with CHC, impairments in insulin action (insulin resistance) and pancreatic -cell failure normally precede diabetes development. In these individuals, the primary sites of insulin action and resistance are muscle and adipose tissue (peripheral compartment) and the liver. With regard to IR in CHC, a number of studies using HOMA-IR (homeostasis model of IR, based on a fasting plasma glucose and insulin level) have found reduced insulin sensitivity in nondiabetic subjects with minimal liver fibrosis;[6] HOMA-B (homeostasis model of cell function, also based on fasting glucose and insulin levels) has not suggested an insulin secretory defect, but this needs more adequate assessment. The association between CHC and IR is of clinical significance to hepatologists, because IR is a predictor of fibrosis progression[7] and a reduced response to antiviral therapy, irrespective of genotype.[8]
Our understanding of the pathophysiology of IR in CHC has been limited by a number of factors. For example, HOMA-IR reflects the fasting state, does not distinguish between peripheral and hepatic IR, and has not been validated in populations with liver disease where insulin clearance rates may be affected. Moreover, factors that contribute to IR such as ethnicity, hepatic steatosis, body fat distribution, and family history of type 2 diabetes have not always been assessed. To date, the pathophysiology of IR in CHC is not well defined, and it is unclear if derangements in insulin action are a direct consequence of the virus or if they arise through indirect mechanisms such as lipid accumulation or cytokine secretion. Finally, there has also been some suggestion that IR in CHC may be genotype-specific, with one study showing lower HOMA-IR scores in genotype 3 infection; the mechanism of any such effect has not been elucidated.[6]
CHC infection results in a chronic inflammatory process in the liver where there is close proximity between hepatocytes, Kupffer cells, and infiltrating cells of the immune system. This inflammatory state with activation of proinflammatory cytokines such as tumor necrosis factor-alpha (TNF-) and interleukin-6 (IL-6), together with the effects of the virus on hepatic lipid metabolism, may inhibit insulin signaling pathways in the liver, increase steatosis, and induce hepatic IR. A hepatic locus of IR is also suggested by a transgenic mouse model of HCV core protein. These transgenic mice develop hepatic IR (prior to development of hepatic steatosis) with elevated intrahepatic TNF- levels.[9] Additionally, in vitro experiments in hepatocytes transfected with HCV proteins demonstrate postreceptor impairments in the insulin signaling cascade, with defects of insulin receptor substrate-1 and insulin receptor substrate-2 content or phosphorylation,[10] and up-regulation of suppressor of cytokine signaling-3.[11] Accumulation of lipid in the liver resulting from virus-induced changes in lipogenic genes[12] and mitochondrial dysfunction[13] has also been proposed to contribute to the inhibition of insulin signaling. Several human studies demonstrate elevated TNF-a levels (in serum and liver) in CHC[14] that may contribute to hepatic IR. However, TNF-a levels have not been strongly correlated with IR and similarly, elevated TNF-a levels are found in other chronic liver diseases such as chronic hepatitis B which are not associated with IR. Other adipokine disturbances in CHC include elevated IL-6 and retinol-binding protein 4,[15] and possibly alterations in adiponectin; these could contribute to hepatic IR, but correlative data does not support their importance.
In this issue of HEPATOLOGY, Vanni et al.[16] have used the gold standard measurement of IR - the hyperinsulinemic-euglycemic clamp with tracer glucose infusion - to more clearly define the site of IR in nonobese, normoglycemic subjects with CHC, compared to controls matched for body mass index and sex. The site of the IR was unexpectedly both peripheral and hepatic. Hepatic glucose production was increased basally and less suppressed with insulin in the subjects with hepatitis C compared to controls. Liver fat and the extent of liver disease were not associated with IR. The authors further suggest that the IR may primarily relate to increased intrahepatic and peripheral fat oxidation and increased hepatic expression of suppressor of cytokine signaling-3 and IL-18. Although this hypothesis is attractive, their data would indicate that a significant component of the IR in the subjects with CHC occurs in the peripheral compartment, although the mechanisms by which this occurs remain elusive (Fig 1). The similar suppression of lipolysis by insulin in all subjects (as measured by glycerol turnover and nonesterified free fatty acids) argues against adipocyte IR and infers that the peripheral IR is predominantly in muscle. However, it should be noted that alterations in lipid oxidation can be a consequence rather than a cause of IR. Of interest, there was no correlation between IR and viral load. A separate analysis of subjects with genotype 3 and subjects infected with other than genotype 3 would have been useful in evaluating whether, as previously suggested, genotype-specific differences occur in CHC-associated IR. Furthermore, such a study would have clarified the relationship between liver fat and IR, because IR is thought to be greater in genotype 1 infection, yet paradoxically, greater hepatic steatosis is present in genotype 3 disease. In the study by Vanni et al., small numbers precluded such an analysis. It should also be noted that liver fat was not significantly different, although the mean level in genotype 3 was 50% greater than in non-genotype 3 subjects.
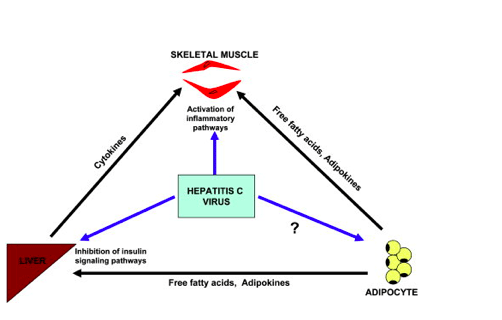
Figure 1. Possible mechanisms by which chronic hepatitis C might cause insulin resistance. The hepatitis C virus is known to affect lipid metabolism and increase cytokine production in the liver, both of which could contribute to hepatic insulin resistance. The mechanisms by which the virus contributes to peripheral insulin resistance are unclear, but might involve direct effects on muscle and indirect effects via free fatty acid flux or adipokine production from adipose tissue.
[Normal View 37K | Magnified View 86K]
What is it then in a viral liver disease that causes skeletal muscle IR? Given that HCV alters lipid metabolism, a simple explanation would be an increased fatty acid supply to muscle, which is a proven cause of muscle IR. This or previous studies have not measured intramyocellular lipid, but the relatively normal circulating fatty acid and glycerol levels and normal suppression with insulin do not lend support for this mechanism. Another likely mediator is via circulating cytokine levels, which were not measured in this study. In this regard, TNF-a and other adipokines including adiponectin, IL-6, and retinol-binding protein 4 might contribute to peripheral IR, but they have not consistently correlated with HOMA-IR measurements.[17] Could the virus affect muscle directly? HCV has a very high circulating load but has not been shown to replicate in muscle.[18] Therefore, one could postulate that a local muscle effect of the virus would likely be mediated by receptors or other molecules on muscle cell membranes, perhaps exciting c-Jun N-terminal kinase-related pathways, or by influencing neighboring cells (e.g., adipocytes) to generate a paracrine effect. Muscle biopsy would not resolve the issue as to whether the virus is present in muscle because of inevitable blood contamination, but might identify activation of pathways known to interfere with insulin signaling. Clearly, whereas much has been learned by this important study, future reports will need to include detailed measurements of lipid compartments, body composition, and more extensive measurements of adipocytokines.
Chronic hepatitis C, although predominantly a liver disease, has protean manifestations. Although the pathophysiology of hepatic injury in CHC is now better understood, that of its other faces is still being elucidated. The study by Vanni et al.[16] significantly increases our understanding of the physiology of IR in CHC. However, like most things in science, their findings raise interesting questions and much food for further study.
|
|
| |
| |
|
|
|