| |
Structural biology: Aerial view of the HIV genome
|
| |
| |
"Structural biologists can now use this genomic map to judiciously zoom in on pieces of the HIV-1 genome and determine architectural and functional principles at the atomic level."
Hashim M. Al-Hashimi1
Nature 460, 696-698 (6 August 2009)
A bird's-eye view of the higher-order structure of HIV-1's entire RNA genome reveals new motifs in surprising places. Structural biologists can now zoom in on these regions to explore their functions further.
The genome of RNA viruses, such as the human immunodeficiency virus (HIV; Fig. 1), folds to form higher-order structures with stems and loops that contain motifs directing various steps of viral replication. Structural biologists usually 'cut out' these motifs and zoom in to determine their three-dimensional structures in an attempt to further understand their function. On page 711 of this issue, however, Watts et al.1 zoom out and provide an 'aerial view' of the secondary structure of the entire HIV-1 genome. Using an innovative technique, they identify functional RNA motifs in surprising regions and define principles that govern the organization of the structure of the HIV-1 genome.
Transmission electron micrograph of a section through HIV. The virus is surrounded by an outer coat (red), and the RNA genome is enclosed in an inner protein core (pink).

The methods commonly used to obtain detailed atomic-resolution images of biomolecules - nuclear magnetic resonance (NMR) spectroscopy and X-ray crystallography - have limitations that preclude analysis of the structure of entire RNA genomes. Both techniques rely on measuring the interactions between light and matter. In NMR spectroscopy, a solution containing the RNA of interest is immersed in a magnetic field and radiofrequencies are used to excite signals from its individual nuclei. As the size of the RNA under analysis increases, the signals become weaker and more congested, limiting structure determination of RNAs that are hundreds of nucleotides long.
X-ray crystallography measures the diffraction of X-rays when they strike crystals containing ordered arrays of RNA. Although the technique does not suffer from size limitations, obtaining ordered crystals for highly flexible and diverse genomic RNA structures is a daunting task. High-resolution structure determination is also a time-consuming process, and as such is reserved for those privileged motifs that have been deemed functionally important. Consequently, more than 80% of the HIV-1 genome remains structurally uncharacterized.
Using a technique called SHAPE (selective 2'-hydroxyl acylation analysed by primer extension), Watts et al.1 provide insight into the complete HIV-1 genome structure. The images produced are of lower resolution than those obtained by NMR spectroscopy and X-ray crystallography, but they span a much larger area of the genome. The technique is thus akin to zooming out on a map and getting a broader view of the landscape at the expense of fine details (Fig. 2).
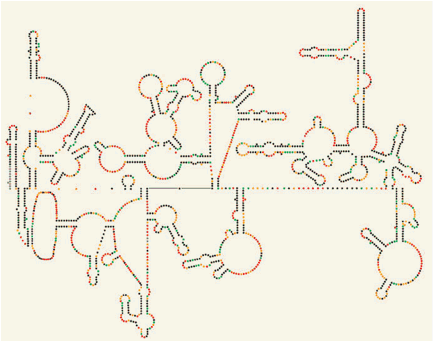
Figure 2: The HIV-1 RNA genome shapes up
Secondary structure of a section of the HIV-1 RNA genome as determined by Watts et al.1 using the SHAPE technique. Nucleotides are represented as coloured dots, with the colours depicting the amount of SHAPE reactivity, which reflects nucleotide flexibility and base pairing. The authors show that the genome has structured motifs (including stems and loops) in regulatory and protein-coding regions. Structured RNA in protein-coding regions may have a role in protein translation and in ensuring correct protein folding. SHAPE analysis of the entire HIV-1 genome is shown in Figure 2 on page 713.
SHAPE exploits the fact that the folds and loops of RNA molecules are stabilized by nucleotide base pairing. The technique relies on interactions between RNA and chemical reagents that react selectively, but sparsely, with flexible (unpaired) nucleotides, and which thereby modify their chemical structures. The reverse transcriptase enzyme is then used to make linear DNA copies of the RNA, which are terminated whenever the enzyme encounters a chemically modified site. By comparing the length and quantity of the DNA copies produced from RNA that has been exposed to chemical reagents with those from RNA that has not, the absolute reactivity, and thus flexibility, of individual nucleotides can be determined. Regions with low reactivity and flexibility correspond to regions of RNA with significant base-paired secondary structure, whereas regions with high reactivity and flexibility correspond to unpaired nucleotides.
Although there are various well-established chemical and enzymatic approaches for probing the secondary structure of RNA2, many of the reagents used react with only a subset of nucleotides (for example, guanine and cytosine versus adenine and uracil) and their reactivity can depend on both RNA secondary structure and the intrinsic nucleotide activity. The unique reagents and chemistry used by SHAPE can help to surmount many of these limitations, and its automation allows for high-throughput studies of very large RNA structures. By plugging the SHAPE reactivity data into a computer algorithm that calculates the thermodynamic stability, and therefore the folded state, of RNA3, 4, Watts et al. propose a model of the secondary structure of the entire HIV-1 RNA genome, which contains a dazzling 9,173 nucleotides.
The structured regions of the HIV-1 genome are concentrated in about 21 large domains1. Most of the functionally important structured RNA motifs that have been characterized so far reside in the untranslated (non-protein coding) region near the ends of the viral genome, which regulates viral replication and packaging of viral particles. Watts and colleagues1 detect these previously characterized RNA motifs, sometimes as components of larger motifs, but they also identify structured RNA elements in protein-coding regions of the genome.
Many HIV-1 proteins are translated into polyprotein precursors by the ribosome as the viral RNA passes through it. The proteins are joined like beads on a string by linker peptides: these are later cleaved to release the individual proteins. There are also unstructured linker peptides between domains that make up the individual HIV proteins. Intriguingly, many of the newly identified structured RNA elements are located in regions that code for these flexible linkers. The authors propose that the structured RNAs slow down protein translation because these regions must be unfolded prior to entry into the ribosome. Because HIV proteins might be folded during translation - a process referred to as co-translational protein folding - this ribosomal pausing may provide additional time for proteins to adopt their correct three-dimensional structures.
This fascinating relationship between RNA and protein structure is not without precedent. The correlation between the secondary structure of messenger RNA and protein translation was recognized as early as three decades ago, and there are several studies showing that mRNA secondary structure can promote ribosomal pausing and modulate other aspects of translation and protein folding5. Watts and colleagues1 go on to identify several other pause sites that seem to buy time at strategic moments during translation. For instance, unwinding folded RNA may allow binding of the signal-recognition particle to the elongating peptide chain. This protein-RNA particle guides the ribosome-peptide-chain complex to the endoplasmic reticulum for further processing. Ribosomal pausing may also provide time for frameshifting - whereby the ribosome stalls and skips over nucleotides without translating them, changing the reading frame - which allows translation of alternative HIV proteins from the same RNA.
On the other hand, highly unstructured regions are observed in hypervariable regions of the HIV-1 genome, which have important roles in viral host evasion. These unstructured regions are bordered by conserved and stable RNA structures that may help to prevent their interaction with the less variable neighbouring regions.
Whenever possible, Watts et al.1 interpreted the details of the SHAPE model in relation to other biochemical and structural data, and information about evolutionary conservation of the pairing possibility of nucleotide regions. But there are still potential sources of error, particularly for an RNA structure of this size. Many regions probably do not exist as a single secondary structure, instead alternating between different conformations. The SHAPE-directed folding algorithm also fails to recognize some RNA structures, such as pseudoknots, or base pairs that form only as part of higher-order tertiary interactions. Atypical RNA structures may also interfere with the SHAPE analysis.
Notwithstanding these limitations, the study by Watts et al.1 is a considerable achievement, showing the feasibility of obtaining 'aerial' views of large genomic RNA structures that reveal their architecture and possible functions. Structural biologists can now use this genomic map to judiciously zoom in on pieces of the HIV-1 genome and determine architectural and functional principles at the atomic level. Bridging these disparate RNA structure-function scales as well as moving towards movies of the genome in functional motion will be challenges for the future. But for now, it seems that the quest for a high-resolution structure of the entire HIV-1 RNA genome has begun in earnest.
|
|
| |
| |
|
|
|