| |
FDA Antivirals Advisory Panel Recommends Maraviroc for Naives Approval 10-4
|
| |
| |
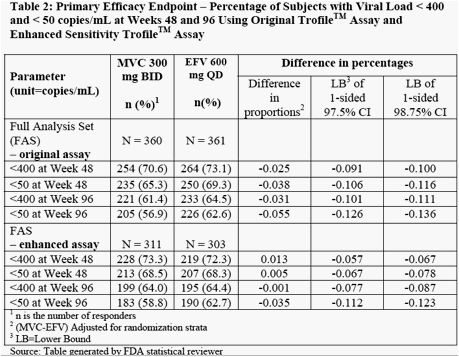
Panelists expressed concern that data at week 96 regarding % <50 was not noninferior but FDA said primary endpoint was 48 weeks, and the panel should be commenting on primary endpoint not what they think it should be. My recollection is that in other drug studies it is not uncommon to see differences out to 96 weeks compared to week 48. Jules
The documents were posted on the FDA's website here
The main reasons considered by those voting no was that in the study discontinuations due to viral efficacy was greater with MVC than EFV (12% vs 4%), although reasons for discontinuations due to adverse events were greater with EFV (11% vs 3%). So the no voters expressed concern about the viral efficacy point, but the yes voters were more concerned that patients should have another option, they think efficacy was good enough, and that safety & tolerability were good for MVC. A number of panelists expressed concern regarding MVC being a preferable Guidelines firstline option, but the FDA said in response we are not voting on whether or not MVC should be a preferred firstline option, only if MVC sould be available as a firstline option.
Jules
QUESTION: Do the safety, efficacy and resistance data presented support the approval of MVC in treatment-naïve HIV-1 infected patients with CCR5-TROPIC virus? If not, please discuss what additional data are needed to provide sufficient evidence of efficacy and safety. If so, please comment on additional data that sponsor should provide post-marketing to further characterize the safety and efficacy profile of MVC i.e.Do the benefits outweigh the risks in this patient population for maraviroc use
VOTE: yes 10, no 4, abstain 0. The panel is now moving on to discussion of last 2 questions
- For future HIV drug development programs, what should be the standards for a treatment naive indication? For example, comment on treatment duration, control arm, margin of efficacy or other study design issues.
- Please discuss the MVC exposure-response data, and whether the magnitude of benefit would justify therapeutic drug monitoring.
VOTING
Neaton no.
Michael Marco no.
Victoria Cargill yes.
Bob Grant yes (lipid profile)
Craig Hendrix yes.
Peter Havens yes. (FDA label should capture tradeoffs between virologic efficacy, safety)
Tracy Swan no. (not overwhelmed with efficacy)
Alice Pau yes. (needs postmarketing studies)
Michelle Roland yes. (need more postmarketing studies)
Barbara McGovern no.
Russell Van Dyke yes. (drug may be more useful in naives due to R5 prevalence early)
Dennis Dixon yes.
Doris Schrader yes. (in the future medicine will be a targeted therapy)
Craig Hegedon yes. (new traget should be available for patients)
Discussion before vote:
FDA says you are not voting on whether this drug should be a preferable choices in Guidelines, only if it should be an option for naives.
Neaton says no, he wants to see another study.
Bob Grant argues for the benefits for having options, its well tolerated and safe & thats important in practice, and greater CD4 increase may provide immune benefits based on signal in study, less TB. I guess thats a yes.
Michael Marco was critical of approval, says no.
Barbara McGovern, says no.
Dr Peter Havens ?,
Dennis Dixon from NIAID/NIH says using primary endpoint week 48 provides adequate evidence that MVC is safe and effective. He is inclined to support MVC for approval.
Enrico Veltri (Schering-Plough) supports approval, its about tradeoffs and options.
Michelle Roland says she is ambivalent but considering what the FDA says the drug doesn't have to be a preferable firstline Guidelines choice but merely should MVC be a firstline option. "ADAPs are going bankrupt". She didn't give yes or no, but it appears she will support.
From Briefing Document FDA
"No safety issues" Howard Mayer, MD, Pfizer, live at FDA hearing
In conclusion, MVC appeared to be well-tolerated in this Phase 3 study in treatment-naïve HIV-1 infected subjects, with relatively few subjects discontinuing for adverse events. No clinically significant imbalance was observed in mortality rate, malignancy, or AIDS-defining illnesses. In addition, analyses of hepatic and CPK-related AEs did not detect a safety signal associated with MVC. Additional analyses pertaining to laboratory values are ongoing and may be presented during the advisory committee meeting if any pertinent results are noted.
2.5 Clinical Safety Results
Analyses of the 96-week safety data for the MVC BID and EFV QD groups are provided in this section. Overall, no new safety signals were identified in association with MVC.
Deaths
A total of 12 deaths were reported up to the Week 96 cut-off, 6 subjects in each treatment group (Table 14). A total of 5 subjects died while on study drug or within 28 days of study drug discontinuation. Of these 5 subjects, 2 were in the MVC group and 3 were in the EFV group. The mortality rate was 1.2 per 100-patient-years for both MVC BID and EFV QD. The causes of death were consistent with what might be expected in this trial population, and were mostly related to underlying HIV infection and/or due to complications of AIDS. There were no additional deaths reported in the periodic safety update reports (PSURs) 1, 2 and 3 covering the period up to February 05, 2009.
Infections and Infestations
As MVC blocks a receptor used by immune cells, there has been concern regarding the potential increased risk of infections due to exposure to this drug. However, no overall increase in infections was observed in association with MVC during Study 1026.
A similar percentage of subjects (62%) reported infections and infestations in each treatment group. However, there was a higher frequency of nasopharyngitis (13% versus 9%) and bronchitis (13% versus 9%) in MVC-treated subjects compared to the EFV group (Table 17). Unlike the phase 3 treatment-experienced trials, no increase in the incidence of upper respiratory tract infection or influenza was noted with MVC. The incidence of sinusitis, gastroenteritis and rhinitis was higher in the EFV group.
Malignancies
Due to the possibility that inhibition of the CCR5 co-receptor could result in decreased immune surveillance, there has been concern regarding the carcinogenic potential of MVC. All malignancies reported during the trial were assessed (Table 15).
There were 18 malignancies (occurring in 16 subjects) reported during the double-blind portion of Study 1026. Thirteen occurred in the EFV group, and 5 in the MVC group. Of these, 12 were considered serious adverse events (4 in the MVC group and 8 in the EFV group). Three malignancies were considered related to the study drug by the investigators, all in the MVC group (nasopharyngeal carcinoma, Hodgkin's disease, and diffuse large B-cell lymphoma). Three subjects in each treatment group were permanently discontinued due to an adverse event of malignancy. Seven subjects died from malignancies, 3 in the MVC group and 4 in the EFV group. Based on these results, there was no increased frequency of malignancies observed in association with MVC administration.
Hepatotoxicity
Patients were excluded from this study if they had transaminases (>3 times the upper limit of normal [ULN]) or total bilirubin (>2 ULN). HBV and HCV co-infected subjects were included in this study. However, subjects with known hepatic cirrhosis were excluded.
A potentially drug-related case of hepatotoxicity resulting in hepatic transplantation occurred in Study 1026 in the MVC QD treatment arm. This subject had alternative explanations for liver disease and had evidence of worsening liver inflammation at the time of starting MVC. In addition, a potentially drug-related case of hepatotoxicity associated with rash, fever and eosinophilia in a healthy subject with concurrent Group A Streptococcal infection was reported in a Phase 1 study (Study 1066). Based primarily on this report in a healthy volunteer, MVC was considered potentially hepatotoxic at the time of its accelerated approval and a Box Warning was placed in the label.
In Study 1026, only 1 subject in MVC 300 mg BID group and 1 subject in EFV 600 mg QD group met the biochemical definition for Hy's law while on study drug. Both cases had an identifiable cause for these abnormalities apart from study drug, including biliary sludge, pancreatitis, alcoholism and hepatitis C.
There were a total of 21 liver-related AEs reported in 19 subjects (Table 16). Six subjects experienced 6 AEs in the MVC BID group, and 13 subjects experienced 15 AEs in the EFV group. Of these, one SAE of hypertransaminasaemia was reported in the MVC group and 1 SAE of hepatitis was reported in EFV group. There was no increase noted in all-causality hepatobiliary AEs or SAEs in the MVC group. The discontinuation rate due to hepatic events was low in both groups (2 in the EFV group and 1 in the MVC group), and there were no fatal outcomes reported related to hepatic events.
CDC Category C Events
No overall increase in Category C AIDS-defining illness was observed in MVC BID group. There were fewer subjects in MVC treatment group compared to the EFV group with Category C events (3.6% vs. 4.9% respectively; Table 18). There were 6 subjects with Category C malignancy in EFV treatment group versus 2 in the MVC treatment group.
Cardiovascular Events
All causality cardiac AEs were reported in 31 subjects under the system organ class (SOC) of cardiac disorders (15 [4.2%] subjects in MVC BID group and 16 [4.4%] subjects in EFV group). The incidence of cardiac events possibly related to coronary heart disease was low in both treatment groups. There were a total of 6 SAEs reported due to cardiovascular events (3 in the MVC BID group and 3 in the EFV group). There were no fatal events reported due to cardiovascular events in either group.
Muscle Disorder Adverse Events
The number of subjects with muscle disorder-related AEs was less in the MVC group compared to the EFV group (9.1% vs. 10.8%, respectively). There was one report of myositis in the MVC 300 mg BID treatment group. This event was Grade 4, considered related to study drug by the investigator and resulted in permanent discontinuation from the study although it was not listed as a serious adverse event. One subject in the MVC 300 mg BID treatment group and 2 subjects in the EFV 600 mg QD treatment group had SAEs of aspartate aminotransferase increased during the study. One subject in the MVC 300 mg BID treatment group and 3 subjects in the EFV 600 mg QD group had AEs of blood creatine phosphokinase increased of Grade 4 severity during the study. There were no fatal events reported due to muscle disorder AEs in either treatment group. One subject experienced rhabdomyolysis in the study in the MVC QD group.
Postural Hypotension
Adverse events consistent with postural hypotension were assessed as this was the dose-limiting AE observed during MVC clinical development. Two (0.6%) subjects in the MVC 300 mg BID treatment group and 8 (2.2%) subjects in the EFV 600 mg QD treatment group had AEs of syncope (Table 21). Two (0.6%) subjects in the EFV 600 mg QD treatment group had AEs of orthostatic hypotension during the study, which were of Grade 1 and 2 in severity. One subject in the EFV 600 mg QD treatment group had an AE of syncope of Grade 3 severity. One subject in the MVC 300 mg BID treatment group had an SAE of syncope which was Grade 4 in severity and resulted in discontinuation from the study. All of the other AEs of syncope were of Grade 1 or 2 in severity and none resulted in permanent discontinuation.
|
|
| |
| |
|
|
|