 |
|
|
| |
72% OF PATIENTS RECEIVING ANA598 IN PHASE II
COMBINATION STUDY WITH INTERFERON AND RIBAVIRIN
ACHIEVE UNDETECTABLE LEVELS OF VIRUS AT WEEK EIGHT
|
| |
| |
Absence of Viral Breakthrough and Favorable Safety Profile Confirm Previous Results
Data Being Presented Today at the 45th Annual EASL Meeting
Anadys press announcement
LINKS: Anadys at the EASL Annual Meeting, Vienna, Austria, April 14-18, 2010
(not a webcast event)
Key Data Slides from ANA598 Late-Breaker Poster
Late-breaker poster presentation on ANA598 Phase II data: April 15, 2010
Poster presentation on ANA598 preclinical data: April 16, 2010
SAN DIEGO, April 15, 2010 -- Anadys Pharmaceuticals, Inc. announced today that 72% of
hepatitis C patients receiving ANA598 400 mg twice daily (bid) plus standard of care (SOC)
achieved undetectable levels of virus at week eight in an ongoing Phase II study, compared to
38% of patients receiving placebo plus SOC.
The preliminary analysis of results through eight weeks also showed that ANA598 400 mg bid plus SOC was well tolerated, with an adverse event profile comparable to SOC alone. As seen before at 200 mg bid, no patient experienced viral breakthrough on ANA598.
"The data for ANA598 400 mg bid through eight weeks demonstrates potent antiviral activity and a favorable safety profile, as was seen previously at 200 mg bid," said Steve Worland, Ph.D., President and CEO of Anadys. "The absence of viral breakthrough in either cohort to date demonstrates that non-nucleosides with superior pharmacokinetics, such as ANA598, can provide a substantial pharmacological barrier to resistance. Coupled with preclinical results that strongly support combinations with other direct antivirals of diverse mechanisms, we are very pleased that the clinical profile to date establishes ANA598 as an attractive agent to advance into Phase IIb development."
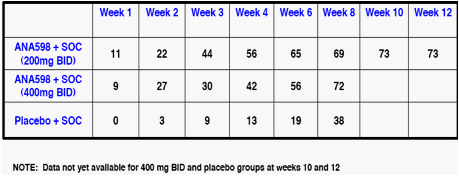
In previously released results for the group receiving ANA598 200 mg bid plus SOC in this study, the high percentage of patients who achieved undetectable levels of virus at week eight (69%) was maintained through week 12, when 73% of patients achieved a complete Early Virological Response, or cEVR, as undetectable level of virus is referred to at 12 weeks. Dosing through 12 weeks in the 400 mg bid group and the control group is currently concluding, and Anadys expects to release antiviral response and safety results through week 12 for these groups in the latter half of May.
Preliminary Antiviral Response Assessment

No patient receiving ANA598 400 mg bid has experienced viral breakthrough (defined as a
confirmed increase of >1 log from any prior measurement) as of the latest data available.
Preliminary Safety Assessment at 400 mg bid
Safety information is available as of a data snapshot that was taken once the last enrolled patient had received eight weeks of treatment. ANA598 400 mg bid demonstrated a favorable safety and tolerability profile through eight weeks, although conclusions regarding safety and tolerability cannot be made until results in more patients and potentially over longer duration are known.
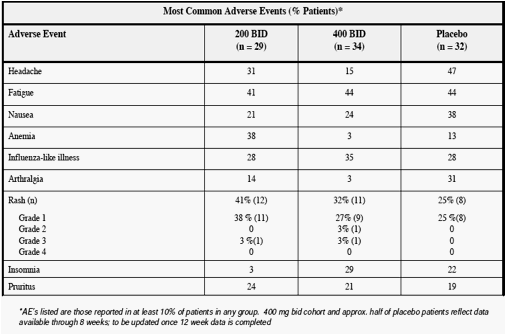
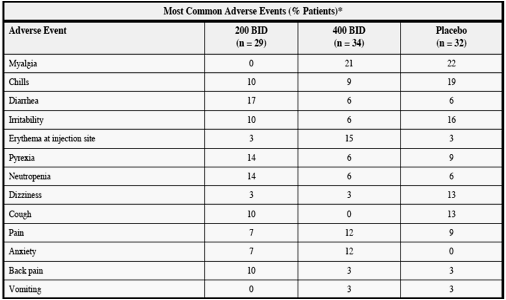
The incidence of all adverse events was similar between the active and control groups, with
reported adverse events being typical for patients treated with interferon and ribavirin. The
incidence of rash was comparable between the ANA598 dose groups and also consistent with
historical reports of rash incidence due to interferon and ribavirin. Through eight weeks, 32% of patients (11/34) receiving ANA598 400 mg bid plus SOC developed rash, compared to 21% of patients at week four and 41% at week 12 for patients who received ANA598 200 mg bid plus SOC. At 400 mg bid, one patient discontinued ANA598 and SOC due to a grade 3 rash and one patient with a grade 1 rash discontinued ANA598. Safety laboratory values were comparable between the ANA598 and control arms.
EASL Presentations on ANA598
The data from the ongoing Phase II study is being presented today in a late-breaker poster
presentation titled "Safety and Antiviral Activity of ANA598 in Combination with Pegylated
Interferon alpha-2A Plus Ribavirin in Treatment-Naïve Genotype 1 Chronic HCV Patients" at the 45th Annual Meeting of the European Association for the Study of the Liver (EASL) in Vienna, Austria. Data through eight weeks is being presented for the group receiving ANA598 400 mg bid plus SOC as well as for the group receiving placebo plus SOC. Data through the entire 12 weeks of ANA598 dosing is being presented for the group that received ANA598 200 mg bid plus SOC.
The late breaker poster and slides of key data excerpted from the poster can be accessed on the Company's website at www.anadyspharma.com.
In addition to the data from the Phase II combination study, Anadys will also present data on the preclinical profile of ANA598 at the EASL meeting on Friday, April 16, 2010. In a poster titled "Enhanced In Vitro Antiviral Activity of ANA598 in Combination with Other Anti-HCV Agents Support Combination Treatment", Anadys will present preclinical data showing enhanced antiviral activity and suppression of resistance when ANA598 is combined in vitro with other anti-HCV agents that act through diverse mechanisms, including protease inhibition, polymerase inhibition (both nucleoside and non-nucleoside inhibitors) and inhibition of host functions. As of
April 16, 2010, the poster will be accessible on the Company's website at
www.anadyspharma.com.
Percentage of Patients with
Undetectable Levels of Virus by Week
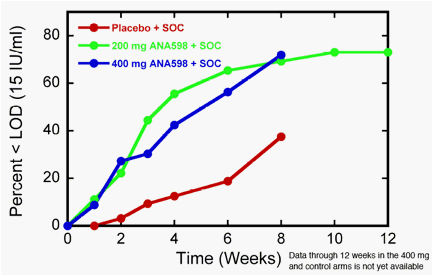
Percentage of Patients with Undetectable Virus (<15
IU/mL) by Study Week
Summary of Reported AEs
from Ongoing Phase II Study*
Phase II Combination Study
In the ongoing Phase II study, a total of approximately 90 treatment-naïve genotype 1 patients are to receive ANA598 or placebo in combination with Pegasys (peginterferon alfa-2a) and Copegus (ribavirin, USP) for 12 weeks at dose levels of 200 mg bid or 400 mg bid, each with a loading dose of 800 mg bid on day one. After week 12, patients are to continue receiving SOC.
Patients who achieve undetectable levels of virus at weeks 4 and 12 will be randomized to stop all treatment at week 24 or 48. The primary endpoint of the study is the proportion of patients who achieve undetectable levels of virus at week 12 (defined as complete Early Virological Response, or cEVR). Additional endpoints include safety and tolerability as well as the proportion of patients with undetectable levels of virus at week 4 (defined as Rapid Virological Response, or RVR). Patients will be followed for 24 weeks after stopping therapy to determine the rate of
Sustained Virological Response, or SVR. Approximately 90 patients have been enrolled in this study - with approximately 30 patients receiving ANA598 plus SOC at each dose level and 30 patients receiving placebo plus SOC. The study is being managed by the Duke Clinical Research Institute (DCRI) under the leadership of John McHutchison, M.D. and is being conducted at a number of clinical sites in the United States.
About ANA598
ANA598 is a non-nucleoside inhibitor of the HCV RNA polymerase and is wholly owned by
Anadys. Anadys has completed three Phase I clinical studies of ANA598 that have demonstrated potent antiviral activity and good tolerability. In a monotherapy study in treatment-naïve genotype 1 patients, treatment with ANA598 for three days led to median end-of-treatment declines in viral load ranging from 2.4 to 2.9 log10 in three separate dose groups. No patient at any dose level showed evidence of viral breakthrough while on ANA598, and there were no serious adverse events. Those patients from the monotherapy study who subsequently received pegylated interferon and ribavirin all exhibited further viral load decline, demonstrating that viral variants revealed by brief treatment with ANA598 remain susceptible to current SOC, consistent with prior in vitro results.
Anadys has completed two long-term chronic toxicology studies of ANA598 (26 weeks duration in rats and 39 weeks duration in monkeys). The No Observed Adverse Effect Level, or NOAEL, is 1000 mg/kg, the highest dose tested, in both the rat and monkey. The completed toxicology studies support the ongoing Phase II clinical study as well as future clinical studies of longer duration.
Anadys has presented in vitro data supporting the use of ANA598 in combination with interferonalpha as well as with direct antivirals currently in development. In particular, data has shown that ANA598 is synergistic in vitro with interferon-alpha as well as representative HCV protease and polymerase inhibitors. In vitro combination treatment at clinically relevant concentrations of interferon-alpha and ANA598 results in clearance of HCV RNA from cells rather than selection of resistant isolates. Furthermore, ANA598 retains full activity in vitro against mutations conferring resistance to protease inhibitors, nucleoside polymerase inhibitors and non-nucleoside polymerase inhibitors that act at binding sites distinct from that of ANA598, while protease and nucleoside polymerase inhibitors retain full activity in vitro against mutations conferring resistance to ANA598.
ANA598 has received Fast Track Status from the FDA for the treatment of chronic hepatitis C.
|
| |
|
|
|
|
|