 |
|
|
| |
VIROLOGICAL RESPONSE, SAFETY, AND PHARMACOKINETIC PROFILE FOLLOWING SINGLE - AND MULTIPLE-DOSE ADMINISTRATION OF
ACH-1625 PROTEASE INHIBITOR TO HEALTHY VOLUNTEERS AND HCV GENOTYPE-1 PATIENTS
|
| |
| |
Reported by Jules Levin
EASL 2010
Victor Detishin1, Wouter Haazen2, Heather Robison3, Lisa Robarge3, Elizabeth Olek3, 1Clinical Hospital of Infectious Diseases and INOPHAR MO S.R.L., Chisinau, Republic of Moldova,
2SGS Life Sciences Services, Clinical Research, 3Achillion Pharmaceuticals, New Haven, CT 06511, USA
CONCLUSIONS
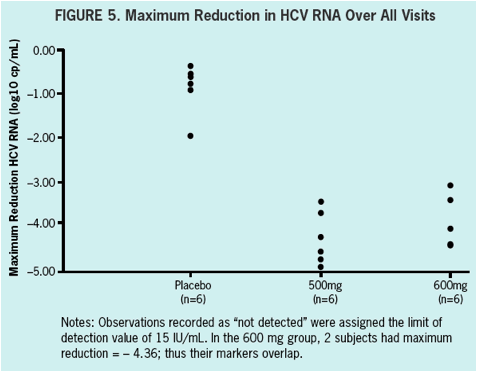
ACH-1625 shows a rapid and robust antiviral effect in genotype 1 naïve and treatment experienced HCV infected patients within the first 48 hours of treatment.
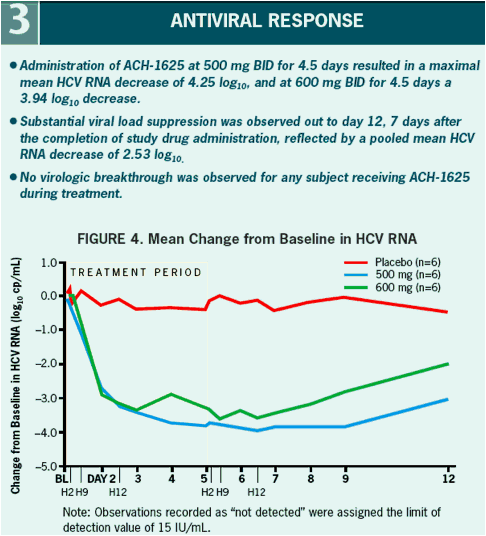
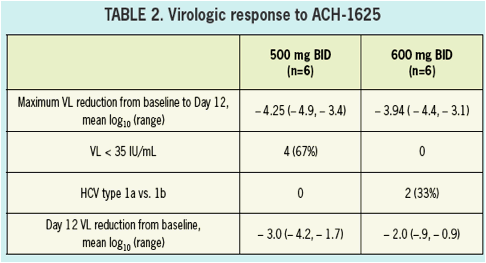
All patients had a more than 3 log10 decrease in plasma HCV RNA with a maximum decrease of 4.25 and 3.94 log10 drop in the 500 mg BID and 600 mg BID dosing groups, respectively.
The sustained viral load suppression observed among patients treated with ACH-1625 at day 12 may yield a potential advantage in longer duration combination treatment.
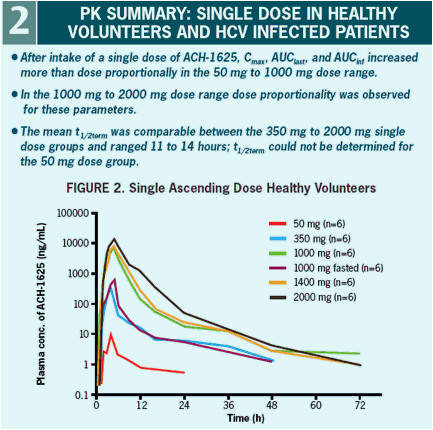
The plasma pharmacokinetic profile shows a dose related increase in Cmax and AUC that is supra-dose proportional at doses below 1000 mg.
ACH-1625 is safe and tolerable at doses up to 2000 mg / day for 4.5 days duration.
Initial results from dosing at 500 mg BID and 600 mg BID indicate that the investigation of additional doses at the QD interval are warranted.
ABSTRACT
Background and Aims: ACH-1625 is a highly potent and specific HCV NS3/4A protease inhibitor (PI). A phase 1 study was conducted to evaluate the safety, tolerability, pharmacokinetics, and antiviral activity following single- and multiple-dose administration of ACH-1625 to male and female healthy volunteers and HCV genotype-1 patients.
Methods: In the single-dose segment 18 healthy subjects received doses of 50-
2000 mg ACH-1625 or placebo in an alternating panel, multiple period, dose escalation
scheme. In the multiple dose segment, 18 healthy subjects received doses of
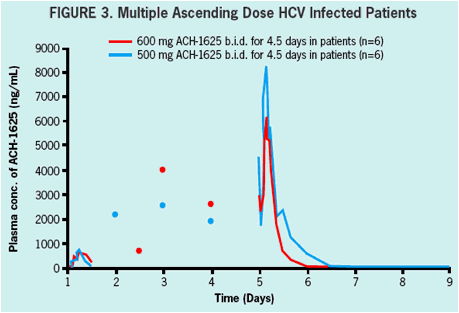
600 mg BID or 1000 mg BID of ACH-1625 or placebo for 5 days in a double-blind
serial-panel study. In addition, 18 HCV patients randomized in a 2:1 ratio received
500 mg BID or 600 mg BID of ACH-1625 or placebo for 5 days in a double-blind serial
panel segment. All patients were followed for 12 days. Clinical safety evaluations
(adverse events, clinical chemistry, hematology, urinalysis, vital signs and ECG) were
performed throughout each segment. Plasma samples for ACH-1625 pharmacokinetic
data were collected. Viral load measurements were taken for the HCV patients.
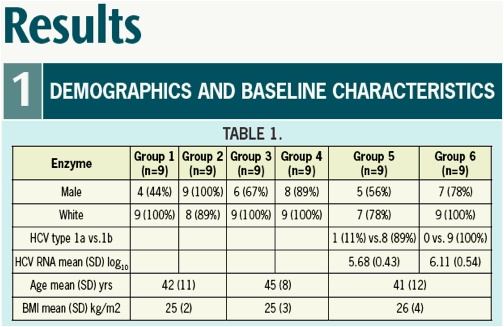
Results: Mean age among patients was 41, BMI 25.9 +/-3.79 SD, and mean log10
VL at baseline was 5.9. A > 3 log10 maximal drop in HCV RNA was observed in all subjects treated with 500 and 600 mg ACH-1625. No response was seen with placebo.
No breakthrough was observed during treatment. Maximal VL decline achieved was
4.25 and 3.94 log10 for 500 mg BID and 600 mg BID, respectively. Two patients in
the 600 mg BID dose group achieved HCV RNA BLQ (15 IU/mL). In patients receiving
ACH-1625 a sustained mean 2.53 log10 decline was observed at Day 12. ACH-1625
showed supra-dose proportional plasma exposure at doses below 1000 mg. There
were no serious AEs reported, nor were there any discontinuations due to AEs.
AEs were mild or moderate and transient.
Conclusions: ACH -1625 is generally safe and well-tolerated, and exhibits plasma
pharmacokinetics and viral load reduction that support once or twice daily dosing.
The sustained viral load suppression observed among patients treated with ACH -1625
at Day 12 suggests a potential advantage that warrants continued investigation.



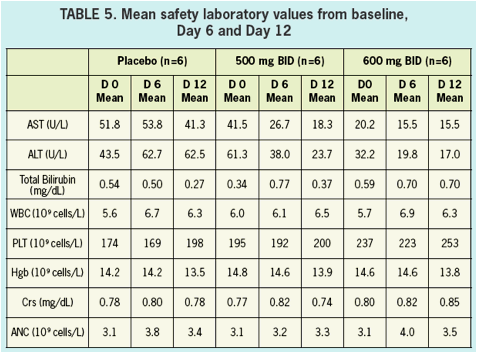
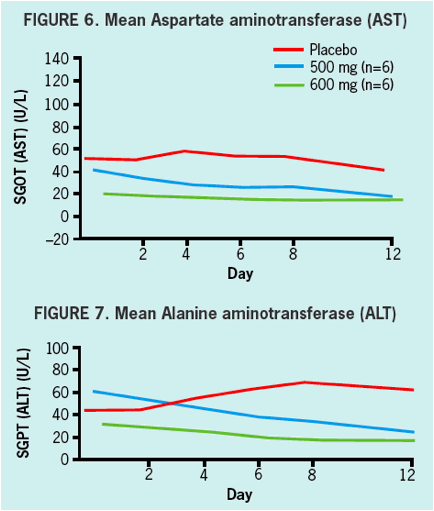
SAFETY & TOLERABILITY
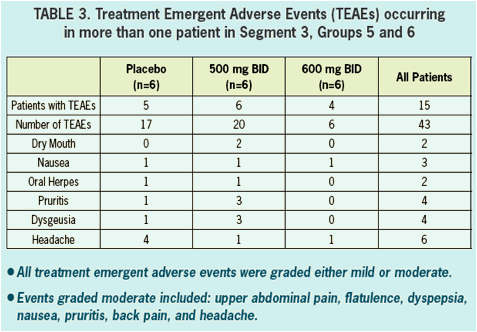
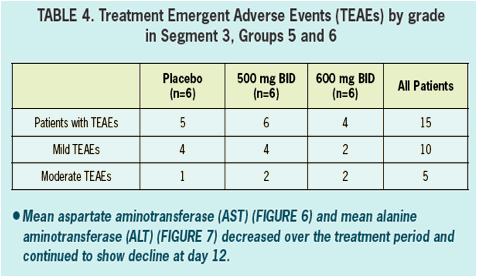
- Safety and tolerability was good in healthy volunteers and in HCV infected patients.
- All adverse events in healthy volunteers were graded mild, and there were no clinically significant changes in vitals signs, ECGs, or laboratory values during treatment.
- There were no serious AEs (SAEs) reported nor were there any discontinuations due to adverse events in either healthy volunteers nor in HCV infected patients.
|
| |
|
|
|
|
|