| |
Long-term use of entecavir in nucleoside-naïve Japanese patients with chronic hepatitis B infection - The current long-term study of entecavir presents results for a cohort of patients treated continuously for 3years.
|
| |
| |
Articles in Press
Jnl of Hepatology March 2010
Osamu Yokosuka1, Koichi Takaguchi2, Shinichi Fujioka3, Michiko Shindo4, Kazuaki Chayama5, Haruhiko Kobashi6, Norio Hayashi7, Chifumi Sato8, Kendo Kiyosawa9, Kyuichi Tanikawa10, Hiroki Ishikawa11, Nobuyuki Masaki11, Taku Seriu11, Masao Omata12
ABSTRACT
Background & Aims
To evaluate the long-term efficacy of entecavir in nucleoside-naïve chronic hepatitis B patients.
Methods
in Phase II studies entered rollover study ETV-060 and received entecavir 0.5mg daily. Responses were evaluated among patients with available samples.
Results
After 96weeks in ETV-060 (120-148weeks total entecavir treatment time), 88% (127/144) of patients had HBV-DNA <400 copies/ml; 90.1% (128/142) had alanine aminotransferase (ALT) ∅1x the upper limit of normal (ULN) among those with abnormal baseline ALT; and 26% (32/121) achieved HBe seroconversion among those HBeAg(+) at baseline. A subset of 66 patients received entecavir 0.5mg (approved dose) from Phase II baseline: at week 96 in ETV-060, 83% (48/58) had HBV-DNA <400 copies/ml, 88% (52/59) had ALT ∅1x ULN, and 20% (10/49) achieved HBe seroconversion. Twenty-one out of 66 patients had paired baseline and on-treatment biopsies: 100% (21/21) and 57% (12/21) demonstrated histologic improvement and improvement in fibrosis, respectively, over 3years. The 3-year cumulative probability of resistance was 3.3% for all patients and 1.7% for the 0.5mg subset.
Conclusions
Long-term entecavir for nucleoside-naïve patients resulted in high rates of virological, biochemical, and histological response, with minimal resistance.
Abbreviations: CHB, chronic hepatitis B, HCC, hepatocellular carcinoma, HBV, hepatitis B virus, HBeAg, hepatitis B e antigen, ALT, alanine aminotransferase, HAI, histologic activity index, ULN, upper limit of normal, PCR, polymerase chain reaction, ITT, intention-to-treat
Introduction
Chronic hepatitis B (CHB) affects 350-400 million people worldwide [1]. The prevalence is highest in the Asia-Pacific region, where 75% of all chronically infected individuals live and up to 25% of CHB patients die of liver cirrhosis, hepatic decompensation or hepatocellular carcinoma (HCC) [2]. In Japan, the prevalence of CHB ranges from 0.8% to 4%, with geographic variation within the country [2], [3], [4], [5]. The vast majority of CHB patients in Japan are infected with hepatitis B virus (HBV) of genotype C [6], [7]. Infection with genotype C virus has been associated with delayed HBeAg seroconversion, more advanced liver disease, and increased probability of HCC development [8], [9], [10], [11].
Recent studies have shown that CHB patients with moderate or elevated serum HBV-DNA are at highest risk of developing long-term complications, including cirrhosis and HCC [11], [12], [13], [14]. Yuen et al. showed that among Asian patients with CHB, disease progression was also seen in patients with persistently detectable viraemia and normal or minimally elevated levels of alanine aminotransferase (ALT), including patients who had achieved HBe seroconversion [12]. Consistent with these findings, current CHB treatment recommendations emphasize the importance of prolonged maximal HBV-DNA suppression and the avoidance of resistance [15], [16], [17].
Medications currently used for CHB include interferons (conventional and pegylated), lamivudine, adefovir, telbivudine, and entecavir. The interferons are efficacious in a subgroup of patients with genotype A infection, low baseline viral load and elevated baseline ALT but are often associated with treatment-limiting adverse events [18], [19], [20]. Lamivudine is well tolerated and initially efficacious, but the emergence of resistance in approximately 70% of patients after 4-5years limits its benefit during long-term therapy [21], [22]. Adefovir treatment is frequently associated with suboptimal HBV-DNA suppression and a cumulative probability of resistance of 29% at 5years among HBeAg(-) patients, and resistance appears to be higher in the HBeAg(+) population [23], [24], [25]. Treatment with telbivudine leads to virological breakthrough, with resistance in 21.6% of HBeAg(+) and 8.6% of HBeAg(-) patients after only 2years [26].
Entecavir has been shown to be highly effective at suppressing HBV-DNA replication to undetectable levels and normalizing ALT in Phase II studies of nucleoside-naïve CHB patients in Japan and in multinational studies [27], [28], [29], [30]. Treatment for 24weeks in the Japanese study ETV-047 showed that entecavir 0.5mg daily resulted in superior viral load reduction compared with lamivudine 100mg daily [28]. In the Japanese study ETV-053, treatment with entecavir 0.5mg daily for 52weeks resulted in significant histological improvement as well as viral load reduction and ALT normalization [27]. Immediately after completion of treatment in study ETV-047 or ETV-053, patients were eligible to enrol in rollover study ETV-060 and receive entecavir 0.5mg daily. We present the long-term efficacy, safety, and resistance results for patients treated with entecavir in Phase II studies who rolled over into study ETV-060, for a total entecavir treatment time of up to 3years (120-148weeks). A subset of patients received the approved dose of entecavir (0.5mg daily) continuously from Phase II baseline, and results for that cohort are also presented.
Discussion
The current long-term study of entecavir presents results for a cohort of patients treated continuously for 3years. The strengths of this study include its focus on a well-defined cohort followed closely over 3years and long-term follow-up liver biopsies on a subset of that cohort enabling a direct assessment of the effect of entecavir therapy on liver disease progression. These results show that long-term treatment with entecavir is well tolerated and achieves histological improvement, durable HBV-DNA suppression, and minimal resistance. Of 167 patients in the cohort, 86% (144) completed 96weeks in the follow-up study for a total of 2.5-3years of entecavir therapy, and only one patient discontinued treatment due to resistance emergence. In both global long-term studies of entecavir and in the present study, continuation of therapy beyond 2years resulted in approximately 90% of patients achieving or maintaining HBV-DNA levels below the PCR assay limit of detection of 300-400 copies/ml [32]. These results were consistent with the results of a sensitivity analysis (last observation carried forward), in which 85% of patients achieved HBV-DNA <400 copies/ml on their last HBV-DNA observation. This method accounts for patient drop-out and missing samples, both of which are common occurrences in long-term studies. However, the interpretation of this sensitivity analysis should be approached cautiously, as it assumes: (1) that subjects who discontinued treatment without achieving HBV-DNA <300 copies/ml would not have achieved it with longer treatment; (2) and that patients who achieved this end point prior to discontinuing would have maintained it over time.
The degree of viral suppression reported in this study is higher than that reported for a cohort of HBeAg(+) patients treated with lamivudine for 3years [33] and higher than that reported for cohorts of HBeAg(+) or HBeAg(-) patients treated with adefovir for 3years [23], [24]. In the current study, 84% of patients were HBeAg(+), and mean baseline HBV-DNA was 7.88log10 copies/ml, 1log higher than the baseline viral load in the adefovir study of HBeAg(-) patients. The rate of HBe seroconversion following entecavir treatment for 3years in this study (26%), is somewhat lower than previously reported for patients treated with adefovir or lamivudine for 3years (40% and 43%, respectively) [24], [33]. This may be related to the large proportion (92%) of HBV genotype C patients enrolled in this study, which has previously been associated with delayed HBe seroconversion [10], [34].
The results of long-term epidemiological-outcome studies have demonstrated that CHB patients with persistently detectable HBV-DNA are at highest risk of liver disease progression [12], [13], [14]. This suggests that long-term suppression of HBV-DNA should help minimize CHB complications. Liaw et al. demonstrated the value of antiviral therapy in a landmark study of CHB patients with cirrhosis or advanced fibrosis treated with long-term lamivudine [35]. Lamivudine-treated patients experienced lower rates of liver disease progression and HCC compared with those who received placebo, but the benefits were reduced by the emergence of lamivudine resistance.
High rates of histological improvement and improvement in fibrosis were observed in the current study among patients who received entecavir 0.5mg from baseline. This improvement in liver histology is likely related to effective viral suppression. Long-term suppression of HBV-DNA is a key objective of CHB therapy, with the ultimate aim of preventing or reversing liver disease progression [15], [36]. In previous studies, maintenance of virological suppression has been associated with improved liver histology among patients treated with nucleoside antivirals. Dienstag et al. showed that long-term treatment with lamivudine resulted in histological improvement, including reversal of fibrosis and cirrhosis; however, those benefits were lost when lamivudine resistance emerged [37]. Mommeja-Marin et al. showed statistically significant correlations between viral load suppression and histological improvement among HBeAg(+) patients treated with nucleoside analogues [38]. Hadziyannis et al. showed that 5years of adefovir therapy for a cohort of HBeAg(-) patients resulted in virological suppression along with improvements in necroinflammation and fibrosis [23]. The current study demonstrates that continued entecavir treatment beyond 1year results in increasing proportions of patients achieving HBV-DNA reduction to <400 copies/ml and further improvements in necroinflammation and fibrosis. At 3years, all patients in the entecavir 0.5mg cohort with evaluable biopsy pairs demonstrated histological improvement, and most (57%) showed improvement in fibrosis, including 85% (11/13) of those who had advanced fibrosis or cirrhosis at baseline.
The potent HBV-DNA suppression achieved in the current study, in combination with entecavir's high genetic barrier to resistance, likely contributed to the observed low rate of resistance emergence: 3-year cumulative probability of resistance of 3.3% for all patients and 1.7% for patients who received the approved dose of entecavir (0.5mg) throughout the treatment period. The rate of 1.7% for patients treated continuously with the approved dose is consistent with that reported in entecavir global studies, in which the cumulative probability of resistance in nucleoside-naïve patients was 1.2% through 5years [39]. The current study differs from the global studies in its focus on a well-defined cohort who were followed continuously with no dose interruption. In comparison with the consistently low rate of entecavir resistance observed among nucleoside-naïve patients, adefovir resistance emerged at rates of 20% among HBeAg(+) patients treated for 5years (median of 235weeks) and 29% among HBeAg(-) patients treated for 5years [23], [24].
In summary, the long-term data presented in the current report demonstrate that continuous entecavir therapy for 3years is well tolerated in Japanese patients and provides durable clinical benefit. The high antiviral potency and low rate of resistance emergence shown in the current study support entecavir as an appropriate choice of first-line therapy for nucleoside-naïve chronic hepatitis B.
Results
One hundred and sixty-seven patients were treated with entecavir in Phase II studies ETV-047 or -053 and entered ETV-060 (Fig. 1). Twenty-three patients discontinued treatment during ETV-060 for the following reasons: adverse event (6), protocol violation (2), withdrawal of consent (4), pregnancy (1), failure to follow-up (4), insufficient effect (1), and complete response (4) or stability of disease condition (1) in the judgement of the investigator. Table 1 shows the baseline (pre-treatment) demographics and disease characteristics for all treated patients (n=167); the cohort of patients who received the approved dose of entecavir (0.5mg daily) from Phase II baseline through the end of treatment (n=66); and for the subset of patients who received 0.5mg entecavir and had biopsies at baseline, week 48 and weeks 144-148. Among all treated patients, 72% were male, and the mean age was 43years. Mean HBV-DNA was 7.88log10 copies/ml, mean ALT was 151IU/l, and 84% (141/167) of patients were HBeAg(+). Ninety-two per cent (154/167) of patients were infected with HBV genotype C. Baseline demographics and disease characteristics were similar for all patient cohorts.
Virological response
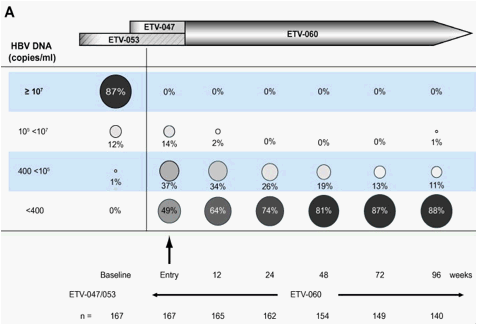
Mean HBV-DNA levels fell rapidly during studies ETV-047 and ETV-053 [27], [28]. For the cohort that entered ETV-060 from the two Phase II studies (n=167), HBV-DNA fell from a mean of 7.88log copies/ml at pre-treatment baseline to a mean of 3.41log10 copies/ml at ETV-060 baseline. Viral load was further suppressed during treatment in ETV-060 and was maintained at low levels through 96weeks (120-148weeks total entecavir treatment time). Forty-nine per cent (82/167) of patients in the cohort had HBV-DNA <400 copies/ml at ETV-060 entry (Fig. 2A). By week 96 of the study, this proportion had increased to 88% (127/144). Of the 82 patients with HBV-DNA <400 copies/ml at ETV-060 entry, 81 patients (99%) maintained this response to the end of treatment. Eighty-five patients had HBV-DNA >400 copies/ml at ETV-060 entry; 62 (73%) achieved HBV-DNA <400 copies/ml during treatment in ETV-060, and 23 (27%) maintained >400 copies/ml at end of treatment. Among the 23 patients who discontinued treatment during ETV-060, 14 had HBV-DNA <400 copies/ml at the last on-treatment measurement. A sensitivity analysis using the last observation carried forward method was conducted based on the intention-to-treat (ITT) population. The last observed HBV-DNA levels for all subjects who either were still on study but had a missing PCR test at week 96 or discontinued prior to week 96 were carried forward; this maintained the total number of subjects in this cohort intact (n=167). When the HBV-DNA end point was re-calculated using this method, 85% (142/167) of patients had HBV-DNA <300 copies/ml at week 96.
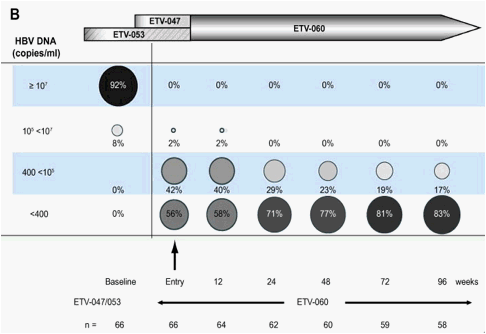
Fig. 2. Distribution of HBV-DNA levels over 96weeks of treatment in rollover study ETV-060 (total entecavir treatment time, 120-148weeks) for (A) the entecavir nucleoside-naïve long-term treatment cohort and (B) the entecavir 0.5mg cohort.
Biochemical response
Almost all patients (97.6%; 163/167) in the Phase II studies had abnormal ALT (ALT >1.0x ULN) at pre-treatment baseline (Table 1 and Fig. 3A). At the time of entry into study ETV-060, 81.0% (132/163) of those patients demonstrated normalized ALT levels (Fig. 3A). By ETV-060 week 48, that proportion had risen to 86.7%, and by week 96 (120-148weeks total entecavir treatment time), the rate of ALT normalization was 90.1%.
Serological response
One hundred and forty-one patients (84%) were HBeAg(+) at pre-treatment baseline (Table 1 and Fig. 4A). At the time of entry into study ETV-060, 16.3% (23/141) of those patients had lost HBeAg and undergone HBe seroconversion (Fig. 4A). By week 96 of ETV-060 (120-148weeks total entecavir treatment time), 38.8% (47/121) of patients had lost HBeAg, and 26.4% (32/121) had undergone HBe seroconversion. Among patients who underwent HBe seroconversion in ETV-060, the majority had achieved HBV-DNA suppression (<400 copies/ml) during treatment in study ETV-047 or ETV-053. One patient lost HBsAg and one patient underwent HBs seroconversion during treatment in study ETV-060.
Resistance
One hundred and sixty-four of 167 patients were monitored for resistance through the end of treatment in ETV-060 (three patients refused consent for resistance testing). Five patients developed genotypic resistance to entecavir, which emerged during the third year of treatment, for a 3-year cumulative probability of resistance of 3.3%. Four of these five patients had received the lower (non-approved) doses of entecavir (0.01mg or 0.1mg) during the Phase II studies prior to ETV-060. Of the five patients with resistance, one patient had achieved HBV-DNA levels <400 copies/ml prior to developing resistance, and four patients experienced virological breakthrough. Fig. 5 provides HBV-DNA and ALT profiles for the patient who received continuous treatment with the approved 0.5mg dose. This patient had detectable levels of HBV-DNA after 48weeks of entecavir treatment in ETV-060. Genotypic resistance testing did not reveal any mutations associated with resistance to entecavir. The patient experienced virological breakthrough at week 96, which was associated with development of entecavir resistance (rt L180M, rt S202G, rt M204V).
Safety
Mean exposure to entecavir during study ETV-060 was 103.9weeks (range: 5.1-140.6weeks). Adverse events were reported for 99% (166/167) of patients, and most were mild to moderate in severity (Table 2). The most common clinical adverse event was nasopharyngitis (16.1%). Increased serum lactic acid (44.3%) and increased lipase (32.3%) were the most common laboratory adverse events. The most common Grade 3-4 adverse event (clinical or laboratory) was increased lipase, which occurred in 6% of patients. The frequency of clinical or laboratory serious adverse events was 13.7% (22/167), the majority of which resolved on continued entecavir treatment. Five patients (3%) discontinued treatment due to adverse events. There were no ALT flares. No deaths were reported during the study.
Entecavir 0.5mg cohort
A subset of 66 patients (66/167) received the approved dose of entecavir (0.5mg daily) from Phase II baseline through to the end of ETV-060. For this subset, among patients with available samples, 83% (48/58) had HBV-DNA <400 copies/ml by week 96 (Fig. 2B). When this end point was re-calculated using the last observation carried forward analysis, 80% (53/66) achieved HBV-DNA <400 copies/ml. By week 96 in ETV-060, 88% (52/59) of patients in the 0.5mg cohort had ALT ∅1.0x ULN (Fig. 3B), 37% (18/49) had lost HBeAg, and 20% (10/49) achieved HBe seroconversion (Fig. 4B). The mean change in HBV-DNA from pre-treatment baseline through to the end of ETV-060 was -5.19log10 copies/ml. Resistance emerged in only one patient in this cohort, for a cumulative 3-year probability of resistance of 1.7%.
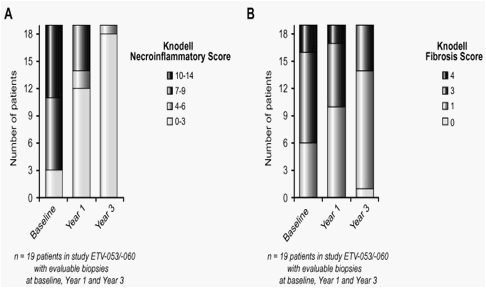
Twenty-one (21/66) patients in the 0.5mg cohort, all originating from study ETV-053, had paired evaluable liver biopsies from pre-treatment (Phase II) baseline and either week 100 or week 148 (ETV-060weeks 48 or 96, respectively). Nineteen (19/21) patients had evaluable biopsies from three time points: baseline, week 48, and week 148. Among this latter subset, 89% (17/19) had HBV-DNA <400 copies/ml at week 148. Histological improvement was observed in 100% (19/19) of these patients from baseline through week 148. There was a marked improvement in the distribution of Knodell necroinflammatory scores with increasing treatment time (Fig. 6A). The two patients who had repeat biopsies at week 100 (but not at week 148) also demonstrated histological improvement from baseline through to week 100. The mean Knodell necroinflammatory score improved from 8.95 at baseline to 1.89 at week 148, and 95% of patients (18/19) exhibited minimal necroinflammation (Knodell NI score ∅3 points) at week 148 (Fig. 6A). Improvements in Knodell fibrosis scores were demonstrated in 63% (12/19) of patients with evaluable biopsies at baseline, week 48, and week 148 (Fig. 6B). Ten patients in this cohort had advanced fibrosis (Knodell fibrosis score=3), and three patients had cirrhosis (Knodell fibrosis score=4) at pre-treatment baseline, and 11 of these 13 patients (85%) showed improvement at week 148. Among 21 patients with biopsies at baseline and either week 100 or week 148, 12/21 (57%) demonstrated an improvement in Knodell fibrosis scores, and 9/21 showed no change. The mean Knodell fibrosis score improved from 2.53 at baseline to 1.47 at week 148. Assessment of liver histology by the New Inuyama classification system confirmed the results obtained using the Knodell classification system (data not shown).
Fig. 6. (A) Distribution of Knodell necroinflammatory scores at pre-treatment baseline, year 1 (48weeks), and year 3 (148weeks), for 19 patients in the entecavir 0.5mg cohort with evaluable liver biopsies at all three time points. (B) Distribution of Knodell fibrosis scores at pre-treatment baseline, year 1 (48weeks), and year 3 (148weeks), for 19 patients in the entecavir 0.5mg cohort with evaluable liver biopsies at all three time points.
Patients and methods
Study design
Study ETV-060 was a rollover study designed to provide open-label entecavir to patients who completed previous entecavir therapy in Phase II studies ETV-047 or ETV-053 in Japan. In study ETV-047, 137 nucleoside-naïve patients were randomized to a range of daily doses of entecavir (0.01mg [n=35], 0.1mg [n=34], 0.5mg [n=34]) or lamivudine 100mg [n=34] for 24weeks [34]). In study ETV-053, 66 nucleoside-naïve patients were randomized to entecavir 0.1mg (n=32) or entecavir 0.5mg (n=34) daily for 52weeks [27]. Patients who completed 24weeks of entecavir treatment in study ETV-047 (n=101) or 52weeks of entecavir treatment in study ETV-053 (n=66) enrolled in ETV-060 and received entecavir 0.5mg daily in an open-label fashion. After 96weeks of treatment in study ETV-060, patients could discontinue the study and were eligible to receive commercially available entecavir, which was approved by Japanese health authorities while study ETV-060 was ongoing. The current analysis describes results for patients who completed 96weeks in study ETV-060 for a total entecavir treatment time of 120weeks (patients from -047) or 148weeks (patients from -053) (Fig. 1). Patients began dosing in ETV-060 immediately after completion of the previous study with no treatment gap or interruption.
During study ETV-060, clinical and laboratory assessments (serum chemistries, haematology, prothrombin time/INR, urinalysis) were made at baseline, at weeks 2 and 4, and every 4weeks thereafter during dosing. Assessments of HBV-DNA by PCR assay and HBV serologies were performed at baseline, weeks 12 and 24, and subsequently every 24weeks during dosing. Baseline liver biopsies in study ETV-053 were performed within 6weeks of initiation of study therapy; or if a liver biopsy had been previously obtained within 52weeks before initiation of protocol therapy, it was used as the baseline specimen for histological evaluation. Liver biopsies were evaluated using the Knodell Histologic Activity Index (HAI) and Knodell fibrosis scores and the New Inuyama classifications [31].
The study was conducted in compliance with the ethical principles of the Declaration of Helsinki, Good Clinical Practice guidelines, and Articles/Notifications of the Ministry of Health, Labour and Welfare in Japan. Written informed consent was obtained from all patients.
Study population
Inclusion criteria for studies ETV-047 and ETV-053 have been described previously [27], [28]. Eligible patients were adults with CHB infection, compensated liver disease, and no more than 12weeks prior treatment with anti-HBV nucleoside analogues. Patients could be HBeAg(+) or (-), and were required to have elevated ALT (1.25-10x the upper limit of normal [ULN] in ETV-047 and 1.3-10x ULN in ETV-053 at screening) and active viral replication (HBV-DNA >105copies/ml by PCR assay at screening in ETV-053 and >107.6copies/ml for patients in ETV-047). Patients were excluded from studies -047 and -053 if they had cirrhosis or evidence of liver decompensation, other forms of liver disease or suspected hepatic tumours, HIV infection or treatment with immunosuppressive therapy or interferon within 24weeks prior to initiation of study medication. Pregnant and nursing women were also excluded.
Efficacy analyses
Efficacy end points included proportions of patients achieving the following: HBV-DNA <400 copies/ml, ALT normalization (ALT ∅1.0x ULN) among patients with abnormal ALT at baseline, and HBeAg loss and HBe seroconversion among patients HBeAg(+) at baseline. Histological end points are presented for the cohort that received entecavir 0.5mg daily from Phase II baseline and include improvement in Knodell HAI and Knodell fibrosis scores among patients with evaluable biopsy pairs. Histological improvement was defined as a >2-point decrease in the Knodell necroinflammatory score and no worsening of fibrosis (worsening: >1-point increase in the Knodell fibrosis score). Improvement in fibrosis was defined as a >1-point decrease in the Knodell fibrosis score. Histological results were also assessed by the New Inuyama classification [31].
Safety analyses
Safety analyses included the incidence of adverse events, serious adverse events, laboratory abnormalities and discontinuations due to adverse events on treatment during ETV-060, including data for patients treated beyond 96weeks. On-treatment ALT flares were defined as ALT >2x baseline and >10x ULN.
Resistance monitoring
During treatment, HBV polymerase/reverse transcriptase substitutions were analyzed for all patients who had HBV-DNA >400 copies/mL at weeks 100 and 120 (from Phase II [pre-treatment] baseline) for patients originating in study ETV-047 and at weeks 100 and 148 for patients originating in study ETV-053. Samples from all patients who experienced virological breakthrough during ETV-060 (increase in HBV-DNA of >1log10 copies/mL from nadir in two consecutive measurements) were also analyzed for HBV polymerase/reverse transcriptase substitutions.
Assay methods
Serum HBV-DNA was determined by Roche Amplicor PCR assay (LOQ=400 copies/mL; Roche Diagnostics K.K., Tokyo, Japan) in a central laboratory. Clinical laboratory tests, PCR assays for HBV-DNA, and serological tests were performed at SRL, Inc. (Tokyo, Japan), the central clinical laboratory designated by the trial sponsor. Genotypic analysis of HBV strains was performed using a PCR-based restriction fragment length polymorphism assay (SRL, Inc., Tokyo, Japan). On-treatment testing for resistance was carried out using a direct-sequencing PCR method.
Statistical analysis
Analyses of efficacy and safety end points were based on patients who received at least one dose of study medication in study ETV-060. Only descriptive summaries were performed. Parameters represented by continuous variables were summarized by the mean, median, standard deviation, minimum, and maximum. Analyses of HBV-DNA as a continuous parameter were applied after log10 transformation. In the analysis of binary end points, patients with missing on-treatment measurements were treated as missing (non-completer=missing). An additional sensitivity analysis using the last observation carried forward method was conducted for the end point of HBV-DNA <400 copies/mL at week 96. In this analysis, the last observed HBV-DNA levels were carried forward for patients without week 96 measurements, i.e., patients who either discontinued prior to week 96 or who were still on study but had a missing HBV-DNA measurement at week 96.
|
|
| |
| |
|
|
|