| |
Lessons From HIV Therapy Applied to Viral Hepatitis
Therapy: Summary of a Workshop
|
| |
| |
"We now stand poised at the threshold of a new era in HCV therapy...>80% SVR rates"....."major future issues....drug resistance"...."Avoidance of resistance is also a regulatory concern". "Currently, there are no standards regarding the evaluation of HCV resistance"..."must....be cautious about the potential for drug interactions".....Open communication between industry, regulatory agencies, clinical investigators, and possibly in a manner reminiscent of the history of HIV therapy, patient advocacy groups should prove helpful in moving the field"
Am J Gastroenterology Jan 19 2010
American Association for the Study of Liver Diseases held a single-topic conference entitled "Viral Hepatitis Therapy: Lessons to be Learned From HIV" on 24-26 July 2008. This article summarizes that conference.
"the ideal drugs would attain >80% response rates with minimal relapse and maximum tolerability"...."As new therapies targeting HCV emerge, limiting or curtailing resistance will become critical, and as important an end point as efficacy, safety, and tolerability. Whatever future HCV triple or even quadruple treatment regimens may look like, the beginning of this era looks bright in terms of achieving higher sustained response rates and curtailing the duration of therapy for our HCV-infected patients."....."One of the major future issues confronting the treatment of HCV with direct-acting antivirals will be drug resistance. Currently, there are no standards regarding the evaluation of HCV resistance-whether it be a standard nomenclature for resistant variants, definitions of virological breakthrough, guidelines for when and how often to test for resistance during therapy, or prediction models for how the HCV virus will react given a certain patient, genotype, viral load, and combination of drugs, akin to advances seen in HIV research." John McHutchison
"Current indications for therapy will likely change significantly with the availability of small molecular inhibitors.....HCV cirrhosis is now a leading cause of death among persons with HIV.....DAA agents (direct antiviral agents) against hepatitis C in coinfected patients raises a number of important considerations: (1) drug-drug interactions with antiretroviral drugs. For example, some HIV PIs inhibit and/or induce cytochrome P450 enzymes, which may increase or decrease levels of agents with similar metabolic pathways (83). Further, many NRTIs require phosphorylation through intracellular kinases to active triphosphate forms, and anti-HCV nucleoside analog polymerase inhibitors may compete with them for phosphorylation (84); (2) HCV drug resistance due to insufficient antiviral activity of PegIFN/RBV; (3) medication toxicity/intolerability (e.g., anemia, rash); and (4) adherence to complex medical regimens targeting both viruses." Mark Sulkowski
"Given the universal interest in studying combinations of DAA agents, the timeline for the development of such combinations is a critical issue. Patients, health-care providers, the pharmaceutical industry, and regulatory agencies are all major stakeholders in this process. Patients and physicians should test combinations with promising potential efficacy at the earliest point that there are preclinical drug interaction, safety, and efficacy studies. Industry must understandably be cautious about the potential for drug interactions or unexpected adverse events that may occur with the use of drugs in combination studies, balancing these considerations with the prospect of taking what would be a momentous leap forward in the field (not to mention the need to remain competitive as others move ahead). Of course, the FDA and other regulatory agencies must put safety as the paramount consideration in evaluating each drug of a proposed regimen before they can be combined. Avoidance of resistance is also a regulatory concern, underscoring the importance of careful observations, including sensitive sequencing studies, in early studies of combination therapy. The pilot study combining a protease and polymerase inhibitor cited above (101) was performed in Australia and New Zealand. It is hoped that such studies will be regarded as feasible in the United States and Europe on the basis of data on each potential component of potential combination regimens gleaned from pre-phase 3 trials. There is a published indication from the FDA Antiviral Products Advisory Committee that this is in accord with the FDA's perspective to a substantial extent (76). Open communication between industry, regulatory agencies, clinical investigators, and possibly in a manner reminiscent of the history of HIV therapy, patient advocacy groups should prove helpful in moving the field in this direction". Ira Jacobson
Alexander Monto MD1,2, Robert T Schooley MD3, Jennifer C Lai MD2, Mark S Sulkowski MD4, Raymond T Chung MD5, Jean-Michel Pawlotsky MD, PhD6,7, John G McHutchison MD8,9 and Ira M Jacobson MD10,11
1. 1Department of Medicine, San Francisco Veterans Affairs Medical Center, San Francisco, California, USA
2. 2Division of Gastroenterology, University of California, San Francisco, California, USA
3. 3Infectious Diseases Division, University of California, San Diego, California, USA
4. 4Department of Medicine, Johns Hopkins University, Baltimore, Maryland, USA
5. 5Gastroenterology Unit, Massachusetts General Hospital, Boston, Massachusetts, USA
6. 6Virology Unit, Hopital Henri Mondor, Universite Paris 12, Creteil, France
7. 7INSERM U841, Creteil, France
8. 8Duke Clinical Research Institute, Durham, North Carolina, USA
9. 9Division of Gastroenterology, Duke University, Durham, North Carolina, USA
10. 10Division of Gastroenterology, New York Hospital, New York, New York, USA
11. 11Department of Medicine, Weill Medical College of Cornell University, New York, New York, USA
Correspondence: Alexander Monto, MD, Department of Medicine, University of California, San Francisco Veterans Affairs Medical Center, 4150 Clement Street #111, San Francisco, California 94121, USA. E-mail: alexander.monto@va.gov
Abstract
Therapies for hepatitis B virus (HBV) have continued to evolve, and new therapies for hepatitis C virus (HCV) will soon be available in clinical practice. These medications for hepatitis C will mark the first time that direct antivirals that target HCV functions have been used. When such drugs are used as single agents, previously existing mutants with reduced susceptibility to them are rapidly selected. The relationship between these drug-resistant mutants and "wild-type" virus is unclear, but resistant strains likely have the potential to maintain the progression of liver disease despite successful treatment of "wild-type" virus. Resistant HBV and now HCV are already a clinical problem. The same issue was recognized very early in the development of therapy against HIV, with azidothymidine-resistant mutants detected within the first weeks of therapy. Clinical investigation and a progressive understanding of the pathogenesis of the disease overcame this challenge and led to the substantial and durable benefits of antiretroviral therapy that are evident today. To bring experts from the fields of HIV and viral hepatitis virology and therapy together for interactive discussions about how to apply the lessons from HIV to the further development of viral hepatitis therapy, the American Association for the Study of Liver Diseases held a single-topic conference entitled "Viral Hepatitis Therapy: Lessons to be Learned From HIV" on 24-26 July 2008. This article summarizes that conference.
HIV BIOLOGY: REPLICATION MECHANISMS AND DYNAMICS, THERAPEUTIC TARGETS, AND RESISTANCE
HIV structure, replication, and mechanisms of inhibition
(Dr Constance Benson, University of California, San Diego)
Mature infectious HIV virions bud from the host cell membrane to form a sphere with an outer lipid bilayer and a nucleocapsid with a dense core. The outer membrane has 72 spiked knobs, which consist of trimers of protein gp120 bound to protein gp4l. Within the virus, two molecules of single-stranded RNA are surrounded by the p17 matrix protein, located between the major capsid protein (p24) that forms the capsid shell and envelope protein, and the p7 nucleoprotein. Viral proteins critical to replication are also incorporated within the virion and include protease, reverse transcriptase, integrase, and others listed in Table 1. Protease is essential for viral assembly; reverse transcriptase and integrase are essential for viral DNA synthesis and integration. Once produced and integrated into the host cell genome, HIV-1 proviral DNA has the genomic structure common to most retroviruses, i.e., gag-pol-env flanked by two long-terminal repeat sequences.
After the viral gp120 membrane protein has bound to the CD4 receptor on susceptible host cells, and to a secondary receptor (one of two chemokine receptors, CCR5 or CXCR4), this complex undergoes a conformational change and the virus enters the cell cytoplasm. Viral disassembly follows using a cellular protein, cyclophilin, which binds to p24. HIV-1 RNA is reverse transcribed to a double-stranded DNA copy and transported to the nucleus in a nucleoprotein preintegration complex in which the viral integrase is responsible for integrating HIV-1 DNA into a host cell chromosome. Integration of viral DNA results in the presence of a linear copy of the viral genome in the host cell. At this point, the virus can remain latent within the host cell indefinitely, with copies being carried to progeny cells with cellular division. With cellular activation through the nuclear factor-κB (NF-κB) pathway, new viral RNA and proteins are then synthesized using host cell enzymes. NF-κB is an inducible transcription factor that splices, caps, polyadenylates, and transports HIV mRNA and genomic RNA transcripts to the cytoplasm where viral regulatory proteins are produced and accumulate, including tat (induces and enhances activity of viral promoter), rev (enhances expression of unspliced and singly spliced mRNAs), vif (induces efficient cell free transmission), vpr (required for nuclear localization), and vpu (enhances virion release from cells). Collectively, these viral proteins greatly accelerate the production of viral gene products and facilitate evasion of restriction factors within the cell that are in place to detect and destroy viruses and other intracellular "intruders" (1).
Over 25 anti-HIV drugs are either available for clinical use or in late stages of investigation. These include agents that inhibit several major steps in the viral life cycle, including viral attachment, reverse transcription, proviral integration, viral protein cleavage, and assembly of mature viral particles (Figure 1). The most numerous and commonly used antiretroviral drugs include nucleoside/nucleotide analogs and nonnucleoside inhibitors of HIV reverse transcriptase (NRTIs and NNRTIs, respectively) and HIV protease inhibitors (PIs) (2).
State of the art: how HIV acquires resistance
(Dr Douglas Richman, University of California, San Diego)
Several common factors contribute to the rapid evolution and generation of genetic diversity that are seen with HIV, hepatitis B virus (HBV), and hepatitis C virus (HCV). These include error-prone polymerases, lack of proof-reading mechanisms, very high magnitudes of replication, and a number of important selection pressures. These selection pressures include drugs, cellular and anatomical compartments (by providing limited space and conditions for replication), and immune responses (3). This rapid evolution and genetic diversity have important implications for the development of antiviral drugs and therapeutic strategies for these chronic infections.
Genetic variants with single and probably double mutations preexist, leading regimens to fail unless a sufficiently high genetic barrier is in place. Genetic HIV variants with three or more drug-resistance mutations rarely exist, however, which is why combination regimens with three drugs are more likely to succeed. The prevention of the cumulative acquisition of drug-resistance mutations requires the sustained suppression of replication (4). Drug resistance can be acquired by transmission (primary resistance), or by the selection of preexisting resistant minor variants during therapy (secondary resistance). The prevention of secondary resistance requires attention to the practical implications mentioned above: patient adherence, provider selection of an optimal regimen and counseling about the importance of adherence, and drug potency, tolerability, and favorable pharmacokinetics.
HIV replication dynamics: implications for HIV therapy
(Dr Ruy Ribeiro, Los Alamos National Laboratory)
HIV viral load in an infected patient remains at similar levels (quasi-steady state) for prolonged periods of time during the asymptomatic stage of infection. However, treatment with potent antiretroviral therapy results in a fast decay of virus in circulation-up to a 2-log decline in as little as 10 days (5,6). This is followed by a much slower phase of decline. The half-life of the second slope of viral decay is on the order of 1 month (7). To explain this slower decline, multiple hypotheses have been considered and modeled, including the existence of long-lived infected cells, viral reservoirs, drug sanctuaries, or immune response effects (7). As the slower late kinetics have been more carefully studied, the previous concept that there was a single second phase rate of decline has become more complex. It seems likely that the slower later kinetics are due to viral replication in a heterogenous group of cells including monocytes, resting lymphocytes, and possibly, other cellular compartments such as astroglial cells. The mean half-life of this compartment seems to be on the order of 4 years, making it unlikely that suppression will lead to viral elimination even if adherence is perfect and the drugs are fully effective (8). This "latent" viral reservoir with slower replication kinetics is primarily responsible for long-term persistence of drug-resistant viral variants that have been selected by earlier failed courses of therapy.
How HIV resistance influences the clinician
(Dr Roy Gulick, Weill-Cornell Medical College)
HIV drug resistance significantly influences the HIV practitioner in at least two distinct patient populations: (i) treatment naive, considering initial treatment options and (ii) treatment experienced, requiring a new regimen because of earlier treatment failure. Approximately 10% of patients newly diagnosed with HIV infection have been infected with viral strains with resistance to at least one of the major HIV drug classes (NRTIs, NNRTIs, or PIs) (9) (Figure 1). Clinical trials data conclusively show that virological failure occurs more frequently on antiretroviral regimens containing drugs to which the virus is resistant. HIV drug-resistance tests, both genotypic and phenotypic, first proved useful when tested in treatment-experienced patients failing a regimen, who went on to subsequent regimens. Drug-resistance testing offered improved treatment management over simply using drug history alone. Testing for genotypic drug resistance has been found to be cost-effective and, as such, HIV-treatment guidelines routinely recommend conducting such testing the first time a patient presents for HIV care and/or before starting antiretroviral therapy (4). Clinicians use this information to select an optimal initial regimen at the appropriate time. They also consider the resistance barrier of the drug regimen (i.e., how many mutations confer resistance to each of the drugs and over what time period), its tolerability, and long-term toxicities among factors that determine the optimal choice of regimens.
The current goal of HIV therapy is suppression of HIV RNA to <50 copies/ml (below the limit of detection) and this is achieved with a regimen containing two, or preferably three, fully active antiretroviral drugs (10). Of particular importance for the heavily treatment-experienced patient are drugs in existing drug classes with demonstrated activity against drug-resistant virus or drugs in newer classes with novel mechanisms of action (e.g., HIV entry inhibitors, HIV integrase inhibitors).
HEPATITIS B: REPLICATION, IN VITRO MODELS, AND RESISTANCE
HBV structure and replication
(Dr Marc Ghany, National Institutes of Health)
Three HBV forms circulate in serum: the Dane particle (intact, infectious virion) and two noninfectious subvirion particles that contain predominantly hepatitis B surface antigen (HBsAg) and host-derived lipids. The viral DNA genome itself is circular and partially double stranded, and the full-length minus strand encodes at least four overlapping reading frames that give rise to four mRNAs, a pregenomic (3.5kb), and three subgenomic (2.4, 2.1, and 0.7kb) transcripts. Functionally important elements for regulation of transcription include promoter elements that are regulated by two enhancer elements (Enl and Enll), a polyadenylation signal within the core gene, and a posttranscriptional regulatory element that is required for efficient processing and transport ( Table 1) (11).
Replication of the HBV genome occurs in the cell cytoplasm within a viral nucleocapsid that consists of the core protein, the pregenomic RNA, and the viral polymerase, which has both reverse transcriptase and RNAse H functions (Figure 2). Nucleocapsid formation requires the coordinated binding of the polymerase to an RNA step loop structure, epsilon, which triggers encapsidation by core particles. The polymerase bound to epsilon serves as a protein primer for DNA synthesis, with epsilon also serving as the template for this reaction. Once the positive DNA strand has been extended from the pregenomic RNA by the HBV reverse transcriptase, a template switch occurs that allows for the circularization of the genome. Once synthesis of the second DNA strand is complete, the viral nucleocapsids can either interact with the envelope proteins in the endoplasmic reticulum to form mature virions that are secreted from the cell, or they can be transported back to the nucleus to replenish the pool of stable nonreplicative forms, the covalently closed, circular DNA (cccDNA), the episomal form of persistence of HBV in cells (12).
Anti-HBV agents, in vitro models
(Dr Scott Bowden, Victorian Infectious Diseases Reference Laboratory)
Current anti-HIV medications were developed and tested using in vitro cell culture systems capable of supporting multiple rounds of HIV replication. Unfortunately, equivalent systems do not exist for HBV, and primary hepatocytes are the only cells able to be infected in vitro. Primary hepatocytes are not suitable for assays that require stringent standardization. The few methods currently used for in vitro testing of anti-HBV agents are labor intensive, technically demanding, and poorly suited to high-throughput screening. Moreover, in vitro studies do not always predict results ultimately seen in vivo (13).
One method of studying HBV replication is the transient transfection of supportive cell lines with well-characterized "laboratory" clones of HBV into which point mutations (e.g., those associated with drug resistance) are introduced by site-directed mutagenesis. The phenotypic effects of specific mutations are deduced by quantifying parameters such as antiviral sensitivity and replication efficiency. Alternatively, phenotypic effects of mutations can be studied in their natural genetic background by transiently transfecting cells with full-length HBV genomes that have been amplified from clinical isolates. Variation in transfection efficiency is the major problem of these methods.
A second method is the tranfection of cells with HBV DNA that is stably integrated into their genomes. This method has been used extensively for screening potential anti-HBV drugs for activity and several sets of cell lines have been created specifically for drug-resistance phenotyping. They carry integrated copies of either a wild-type HBV genome or one of its mutant derivatives, expression of which is controlled by repressible or inducible promoters. New lines have to be created for each HBV mutant and integration of viral DNA into the cellular genome may affect the expression of both viral and cellular genes.
A third method is the transduction of replication-competent HBV genomes into cells that support their replication by means of recombinant baculoviruses (or other viral vectors). This results in a more controllable viral expression than is possible with transfection. The need to continually generate, maintain, and standardize stocks of viral recombinants is the main disadvantage of this approach.
Finally, virtual phenotyping is an approach that is relatively new for HBV, although it has been used extensively to predict phenotypes of HIV isolates. It relies on computer-assisted analysis and correlation of information from large databases of genotypic, phenotypic, and clinical data (14). However, this approach cannot predict phenotypic effects of new mutations.
Mechanisms of hepatitis B virus drug resistance
(Dr Fabien Zoulim, University of Lyon)
Drug resistance is one of the most challenging issues in hepatitis B therapy. The main antivirals available for the treatment of HBV are nucleoside/nucleotide analogs, all of which act as inhibitors of HBV polymerase function. Polymerase gene mutations typically lead to a lower susceptibility of HBV to drugs because of a lower binding activity of the compound in the catalytic site of the enzyme. Viral genome mutations are presumed to be archived in cccDNA (15,16).
Viral replication and turnover are rapid for HBV (see Table 1). Its reverse transcriptase generates multiple quasispecies that coexist and reach population densities in direct proportion to their relative replicative fitness. Hepatocyte lifespan heterogeneity (estimated to be up to 100 days), however, means that therapy for HBV must be prolonged (17). As such, selection of drug-resistance mutations depends on two factors: (1) the relative fitness of mutant viruses in the presence of the drug and (2) the replication space available for the virus, which depends on hepatocyte turnover. Hepatocyte turnover in HBV is slow because the immune-mediated killing of infected cells is slow, providing limited opportunities to replace the original virus population by a new one of drug-resistant variants (18,19). This is one of the reasons why the dynamics of selection of drug resistance are so different between HBV and HIV.
Mutations may lead to decreased susceptibility to the drug, but may also result in reduced replication capacity. Sequential addition of mutations may be required for the virus to achieve higher levels of resistance and better fitness, e.g., for entecavir primary-resistance mutations at reverse transcriptase position 204 (rt204), secondary-resistance mutations at rt184, rt202, or rt250, as shown in Figure 3 (20). Many of the main resistance mutations are associated with compensatory mutations to restore replication capacity. The newer generation of HBV antivirals seems to have a higher barrier to resistance (defined as either (1) a high number of mutations needed for the establishment of resistance and/or (2) a marked reduction in replicative fitness associated with mutant strain, even if it is only caused by a single mutation). The knowledge of the cross-resistance profile of antiviral drugs is also very important in choosing antivirals for add-on combination therapy (21).
Hepatitis B virus polymerase domains (used with permission from ref. 25).
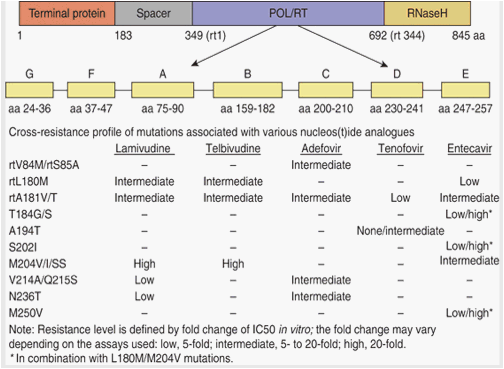
The HBV polymerase has conservatory constraints due to the fact that the HBV genome has overlapping reading frames. Changes at one position in the polymerase may affect the structure and function of the surface protein, as has been shown for some lamivudine-associated mutations (22). Defective or less-infectious mutants may require complementation of mutant proteins with some from wild-type virus to package and propagate (some of the M204I and A181T mutants are examples) (23). Some drug-resistant mutants may escape vaccine-induced anti-HBs antibodies because of mutations in the overlapping surface gene.
There are major clinical implications of these findings. The kinetics of resistant mutant emergence affect the virological monitoring of antiviral treatment (24). Prevention of resistance should rely on first-line therapy using nucleoside analogs exhibiting potent antiviral effects and high genetic barriers to resistance. Second line, add-on strategies should use drugs with a complementary cross-resistance profile.
HBV mutations, alterations in therapy based on kinetics
(Dr Jean-Michel Pawlotsky, University of Paris)
Resistance to HBV reverse transcriptase inhibitors is acquired gradually, through the selection of preexisting resistant variants and gradual accumulation of new amino-acid substitutions that confer stepwise increases in the level of drug resistance. Compensatory mutations may restore the in vivo fitness of the resistant virus, allowing viral replication to return to pretreatment levels (25).
The principal mutations associated with lamivudine resistance are located in domain C, in the YMDD catalytic motif of the HBV reverse transcriptase. They include rtM204V (YVDD sequence) and rtM204I (YIDD), and the more recently identified rtM204S (YSDD), as shown in Figure 3. Additional substitutions that are often coselected with the resistance substitutions at rt204 position, such as rtL180M and rtV173L located in the B domain, can compensate for the loss of replication efficiency of rt204 variants (26). Cross-resistance has been reported between lamivudine and other L-nucleoside analogs, such as telbivudine, emtricitabine, valtorcitabine, clevudine, or elvucitabine, all of which showed significantly reduced antiviral potency on HBV variants bearing the V, I, or S substitutions at position rtM204. In vivo, resistance to telbivudine has been reported to be related to variants bearing an rtM204I (YIDD) substitution only.
Entecavir also shows reduced activity in the face of resistance mutations at rt204, although in vivo, entecavir has activity against lamivudine-resistant variants, but to a lesser extent than does wild-type virus. Additional substitutions have been reported to restore baseline viral replication capacities when associated with substitutions at rt204 (with or without the rtL180M substitution). They include rtT184S/A/I/L, rtS202G/C, and rtM250I//V. Some of them further reduce the entecavir susceptibility of rt204 variants in vitro, whereas they all substantially improve the replicative fitness of rt204 variants in the presence of entecavir. Full resistance to entecavir results from a three-step selection-mutation accumulation process: initially rt204, then rtL180M, which improves the fitness of rt204 variant, and then one or more of the entecavir-specific substitutions that restore full-replication fitness in the presence of the drug, which defines the high genetic barrier of the drug to HBV resistance (27).
Long-term administration of adefovir has been shown to select variants bearing rtN236T or rtA181V/T, both of which confer low-level resistance (5- to 10-fold increase) to adefovir in vitro. The emergence of adefovir resistance is slow and may be associated with different patterns of HBV DNA kinetics, as a result of the low replicative fitness of resistant variants. rtA181V/T substitutions have been reported to confer cross-resistance with lamivudine, and potentially with other L-nucleoside analogs (28). The amino-acid changes that confer resistance to tenofovir administration in vivo are not currently known. rtN236T substitution confers reduced susceptibility to tenofovir in vitro.
The strategy to delay the development of resistance to anti-HBV drugs is to use potent drugs with a high genetic barrier to resistance. Nowadays, only entecavir and tenofovir fulfill these criteria and should be used as first-line antiviral drugs in monotherapy. HBV DNA must become undetectable on treatment in a sensitive real-time PCR-based assay with a lower limit of detection of 10-15lU/ml within the first year of administration. If HBV DNA remains detectable, a second drug should be added with no cross-resistance to prevent the development of resistance.
HEPATITIS C BIOLOGY: REPLICATION MECHANISMS, MECHANISMS OF ACTION OF CURRENT THERAPIES, CURRENT UNDERSTANDING OF HCV RESISTANCE
Mechanisms of replication of HCV
(Dr Brett Lindenbach, Yale University)
HCV is the only member of the hepacivirus genus of Flaviviridae, a family of viruses that has a lifecycle distinct from HIV and HBV. As with other Flaviviridae, the 9.4kb (+) sense RNA genome is translated into a single long polyprotein that is cleaved by both host signal peptidases and virally encoded proteases (NS2, NS3/4A) into 10 functional peptides (structural and nonstructural proteins). One of these proteins, the NS5B RNA-dependent RNA polymerase, catalyzes the direct copying of the viral genome into a replicative intermediate RNA. As there is no DNA intermediate (i.e., no reverse transcriptase activity), HCV is not known to be capable of a latent phase, and is entirely dependent on replication for its lifecycle. Thus, it is not surprising that sustained viral clearance is possible with current antiviral therapy. NS5B is also an error prone and low-fidelity enzyme, and is thus capable of generating the extraordinary sequence diversity described worldwide.
Improved methods of viral cultivation have significantly improved our understanding of the HCV lifecycle. The successful development of HCV replicons, autonomous RNAs the replication of which is directed by the viral replication machinery (nonstructural proteins), has been a major advance not only for the elucidation of HCV RNA replication but also for the screening of candidate antiviral compounds that inhibit replication (29). Studies based on these models have also revealed that HCV replication occurs on altered host membranes derived from the endoplasmic reticulum called membranous webs.
To understand the steps governing HCV entry, the use of pseudotype virus systems, in which the heterologous virus has been engineered to express HCV envelope glycoproteins, has advanced understanding of the receptors and coreceptors participating in replication. Emerging data have identified the requirement for tight junction proteins such as claudin-1 and occludin, as well as CD81, in the entry of HCV into the hepatocyte ( Table 1).
Most recently, the successful ability to support bona fide viral replication in culture (the JFH-1 strain) now allows the entire viral lifecycle to be studied in full context (30,31). Studies using the JFH-1 system have firmly identified a close association between HCV and lipoprotein metabolism. For instance, HCV seems to be secreted from hepatocytes using a VLDL-dependent pathway. Thus, characterization and inhibition of each of the steps in the viral lifecycle are now possible. Still, there remain important limitations to present culture models, including their need for cell lines harboring tissue culture adaptations permissive for viral replication. The development of a tractable animal model remains another important hurdle for the understanding of HCV pathogenesis. However, advances in knowledge of viral entry factors may help to overcome this barrier in the near future.
Mechanisms of action of current antiviral therapies
(Dr Ray Chung, Massachusetts General Hospital)
Current best therapy for chronic HCV is a combination of peginterferon alfa and ribavirin (PegIFN/RBV) (32,33). Interferon (IFN) is a broadly active cytokine and a cardinal component of innate antiviral immunity, which has both antiviral and immunomodulatory properties. IFNs act through the Jak-STAT signaling pathway to upregulate hundreds of so-called IFN-stimulated genes (ISGs). ISG products include some known antiviral proteins, such as protein kinase R and oligoadenylate synthase, which inhibit viral protein and RNA synthesis, respectively. However, the precise repertoire of ISGs that leads to clearance of HCV is as yet unknown. Replicons and infectious HCV virus propagated in tissue culture have been uniformly sensitive to IFN, implying that HCV does not encode IFN resistance. This is not a surprising finding in light of the multifocal and nonsequence-specific nature of IFN's antiviral effects. These findings imply that the observed nonresponse to IFN in clinical populations must be explained by other factors, including, quite possibly, differences in ISG function or expression of IFN antagonists.
The addition of ribavirin (RBV) to regimens has been shown to modestly improve antiviral responses to IFN, particularly among patients with suboptimal responses to IFN. However, the most important clinical effect of RBV is to markedly decrease relapse rates among patients who respond to IFN. Multiple lines of evidence have suggested that RBV acts as an RNA mutagen; in vivo, there seems to be an early but transient increase in RNA species diversity in patients treated with RBV (34). However, more compelling evidence suggests that the addition of RBV to PegIFN may actually enhance ISG expression, thus acting to enhance both the antiviral and, indirectly, immunomodulatory effects of IFN (35). Kinetic data are thus far consistent with such a proposed mechanism, but further study is needed to formally prove this. Ongoing clinical trials in conjunction with HCV protease inhibitors (PIs) have thus far reinforced the indispensability of both IFN and RBV in maximizing sustained response rates.
Advances in solving the crystal structure of the HCV NS3/4A protease and NS5B polymerase led to the development of small molecule inhibitors of these enzymes. Because of the end-product inhibition of protease enzymatic activity, a particularly productive approach has been the design of peptidomimetic inhibitors of NS3/4A. Current compounds in clinical testing, including telaprevir, boceprevir, TMC435350, BI201335, and R7227 are either linear or macrocyclic peptidomimetics. Each has demonstrated robust antiviral activity in replicon models, clinical trials, or both. However, the intensive selection pressure associated with the binding of NS3/4A has been associated with the early selection of resistant variants.
Two different classes of NS5B polymerase inhibitors have emerged. Nucleoside polymerase inhibitors such as R1626 and R7128 bind to the active site of NS5B and produce chain termination of the viral RNA. In at least five other sites, small molecules have been designed to bind to allosteric sites that induce conformational changes in the polymerase. Nucleoside NS5B inhibitors have been shown in some instances to have excellent antiviral efficacy, and a higher barrier to resistance. On the other hand, nonnucleosides have thus far been associated with more limited responses and have a lower barrier to resistance.
Because of resistance concerns with direct-acting antivirals (DAAs), there is an emerging interest in the possible targeting of host factors required for viral replication. Examples of this class of compounds are molecules that target cyclophilin, an identified cofactor for NS5B RNA binding. These agents, which include DEBIO-025 and NIM-811, exhibit moderate antiviral activity with a high barrier to resistance. It is therefore conceivable that the future will bring construction of regimens that combine DAAs with host cofactor inhibitors to minimize or obviate future dependence on IFN-based regimens.
Current understanding of HCV resistance
(Dr David Wyles, University of California, San Diego)
The development of DAAs for HCV, also referred to as specifically targeted antiviral therapy for HCV (STAT-C), has brought new concerns about these agents' selection of resistant strains of virus. Underlying this selection is the error-prone NS5B polymerase, which generates approximately one nucleotide error for each genome replication cycle. Thus, it is not surprising that monotherapy with HCV protease and polymerase inhibitors is associated with rapid selection of preexisting variant species. In replicon assays, these resistant variants replicate with lower fitness than does wild-type virus (36). Evidence to support this finding in vivo is the consistent observation that wild-type quasispecies usually return over a variable period of time after removal of selection pressure. However, with sufficient duration of therapy, it is conceivable that secondary, compensatory mutations could arise that restore fitness to resistant variants. Until a better understanding of these mutations is gained, prolonged exposure to single-agent-targeted therapy should be avoided.
For NS3/4A inhibitors, mutation at the A156 site has been uniformly selected; cross-resistance at this site to all inhibitors has been reported. Broad cross-resistance with mutations at the R155 position has also been reported (37). For nucleoside polymerase inhibitors, mutations in the NS5B active site have been associated with a marked reduction in fitness; thus, the threshold for resistance for this class has been high (38). These patterns include S282T mutations for 2C-methyl compounds, and S96T for 4azido compounds (39,40). Nonnucleoside inhibitors have selected for mutations in each of the four distinct sites targeted for inhibition (41). In general, resistance mutations associated with nonnucleoside inhibitors are unique to the binding site without cross-resistance to inhibitors of other allosteric sites or to active site polymerase inhibitors (42). An exception to this may be the two palm inhibitor binding sites, which lie in close proximity to one another, for which mutations at position 316 have yielded elevated IC50s in vitro to inhibitors of both sites (42).
For host cofactor inhibitors such as cyclophilin antagonists, the barrier to resistance is decidedly higher, but in vitro mutations in NS5A and NS5B have been described (42,43). Encouragement for the design of an IFN-sparing oral cocktail regimen has come from studies describing synergy between two PIs, as well as between PIs and both nucleoside and nonnucleoside polymerase inhibitors (44). Thus, the development of a potent oral antiviral regimen for HCV with nonoverlapping-resistance profiles seems to be theoretically possible.
HCV mutations, replicative fitness
(Dr Tara Kieffer, Vertex Pharmaceuticals)
Every HCV-infected patient carries a heterogeneous population of HCV, including preexisting variants with decreased sensitivity to direct-acting antiviral drugs. The likelihood of the emergence of clinically relevant resistant variants depends on several factors, including the selection pressure applied by the drug, the genetic barrier to resistance, and the replication fitness of the resistant variants. For HIV and HBV, as for HCV, resistant variants are usually not as fit, and this is particularly true if drugs to which they are resistant bind directly to the active site of a viral enzyme. However, a resistant variant can improve fitness by a stepwise accumulation of additional compensatory mutations.
HCV variants with mutations conferring resistance to DAAs have been observed in vitro and in clinical trials. The fitness of these HCV variants is typically estimated in vitro by measuring replication capacity (by transient replication in the replicon system) and enzymatic fitness (by measuring catalytic efficiency) (45,46). DAA-resistant variants have varying degrees of decreased replication capacity compared with wild-type virus. The NS3 A156T mutation, which confers resistance to many PIs, has significantly reduced NS3/4A catalytic efficiency and replication capacity (47,48). The NS5B nucleoside inhibitor-resistant mutation, S282T, also has decreased replication capacity, as illustrated in Figure 4 (46). The nonnucleoside inhibitor-resistant mutation P495A/L has decreased replication capacity, but fitness can be restored by compensatory mutations elsewhere in NS5B. However, because nonnucleoside inhibitors bind to allosteric sites that are not as conserved as the active site, the prevalence of patients with preexisting resistant mutations is higher (49). For many of these DAA-resistant mutations, the decreased replication capacity observed in vitro has not been confirmed in a more physiologically relevant setting. However, recently, the in vivo fitness of viral variants with decreased sensitivity to the HCV PI telaprevir (TVR) has been estimated using a novel method that assessed growth rate in the absence of TVR selective pressure. The replicative fitness of different viral variants was inversely correlated with their degree of resistance to TVR (43).
Fitness of resistant variants is not only important in determining the probability of resistance emerging clinically, but also whether quasispecies dominated by variants will revert to wild type in the absence of drug selective pressure. Resistant variants with significantly impaired fitness will be replaced by wild-type virus more rapidly in the absence of drug selective pressure (Figure 5, ref. (43). The antiviral response in patients whose HCV has reverted to dominant wild type (i.e., resistant variant levels are similar to levels present before therapy) and are reexposed to a DAA is unknown, but will be important in understanding future treatment options for patients who fail initial therapy. Unlike HIV infection, in which resistant variants generated during unsuccessful antiretroviral therapy are archived as latently integrated proviruses in resting memory T cells, no such long-lived reservoir has been demonstrated for HCV. Thus, retreatment with DAAs is a strategy that should be evaluated in future clinical trials (50).
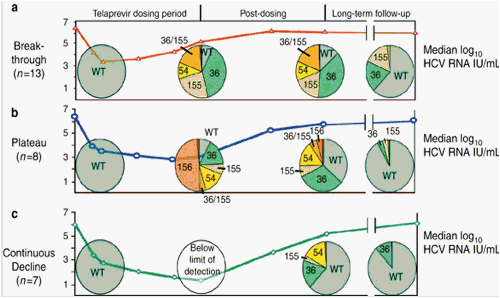
Frequency of NS3 protease variants by response group in three groups of patients treated with 14 days of Telaprevir monotherapy in a phase 1b study. Lines depict mean HCV RNA levels in each group, and pie charts represent the mean frequency of variants from all patients in each group at each time point prior to, at the end of, 7-9 days after, and 2-7 months after treatment. Panel a shows 13 patients who experienced viral breakthrough during treatment, panel b shows 8 patients whose viral load decline reached a plateau during treatment, and panel c shows 7 patients whose viral load continuously declined (used with permission from ref. 43). HCV, hepatitis C virus; WT, wild-type virus.
Will IFN always be required for HCV therapy?
(Dr Alan Perelson, Los Alamos National Laboratories)
Rapid development of drug resistance has been seen when HCV PIs, such as telaprevir, have been used in monotherapy (43). Viral kinetic modeling can provide insight into whether such observations of drug resistance reflect viral adaptation specific to the compounds currently in use or whether they reflect an intrinsic property of all DAA agents. Further, viral kinetic modeling can be used to evaluate whether combinations of DAA agents can be effective in the absence of IFN.
Essentially, all therapies that significantly inhibit HCV so far, including PI monotherapy, show two phases of decline: a rapid decline, referred to as Phase 1, followed by a slower decline, Phase 2. It is accepted that Phase 1 decline comes from the agent substantially blocking viral production, and Phase 2 comes from elimination of infected cells. Compounds such as telaprevir that drop HCV RNA by three logs over 2 days of therapy must shut off viral production almost entirely. For treatment to be entirely effective, however, it needs to proceed until infected cells have been significantly eliminated (i.e., weeks to months for the hepatocyte).
HIV mutations occur during reverse transcription, after which an infected cell contains thousands or more of largely identical viruses. In HCV, a mutation likely occurs with the production of each 1-10 virions (51), and hence preexisting drug-resistant variants are more likely with HCV than with HIV. Given that ∼1012 hepatitis C virions are produced per day, it is likely that all viable single and double mutants are produced each day in each untreated patient. As such, even before therapy with a single DAA, single and double mutants preexist, and hence a drug that requires only one or two substitutions to generate resistance should fail. Taking into account compensatory mutations or decreased fitness of mutants adds additional levels of complexity to such models. The overall findings of such modeling experiments, however, are that combinations of DAA agents with different resistance profiles that can overcome a genetic barrier of three resistance mutations will be needed if we are ever to remove IFN from standard therapy.
THE HIV THERAPEUTIC REVOLUTION: ADVOCACY, "EMERGENCY," NIH FUNDING, ACTG
How HIV achieved co-therapy with multiple experimental agents
(Dr Paul Volberding, University of California, San Francisco)
A panel of the Institute of Medicine convened in 1986 to consider how the new HIV epidemic could be confronted (52). Co-chaired by David Baltimore and Shelton Wolfe, one key and dramatic recommendation was that the government, through the NIH, should invest new funds, not diverted from other research, into the fundamental science of the newly recognized virus. The panel argued against contracts and in favor of investigator-initiated grants as the vehicle for this discovery and anticipated the development of new antiretroviral drugs. They recommended the then startling amount of one billion dollars annually for this new target. The wisdom of these recommendations and their implications for other challenges should, in retrospect, be evident. Even as the panel deliberated, the first candidate drug, azidothymidine was entering clinical trials that would establish its activity. The extension of the real but modest efficacy of that drug absolutely required the basic scientific discovery funded by the NIH investments. The lifecycle of HIV was quickly probed, revealing numerous targets for therapeutic drug discovery. As these candidate drugs became available for human trials, another key NIH investment, in the academically based infrastructure, particularly the AIDS Clinical Trials Group (ACTG), was also essential. A trial element was a mobilized patient advocate community that insisted that the results of basic investigation and drug discovery be considered quickly in human trials and rapidly approved for use. Combination therapy was considered almost immediately, in part because of initial potency limitations but later because of the rapid selection of drug resistance by monotherapy. The success of multiagent cancer chemotherapy also presented a rationale for drug combinations. HIV therapy moved almost immediately from one drug to two-drug combinations. Dual therapy, in retrospect, had considerable clinical efficacy but was quickly eclipsed by the striking benefit of three-drug combinations, especially those including a PI (53,54).
Lessons in drug resistance from HIV-1 drug development
(Dr Daniel Kuritzkes, Brigham & Women's Hospital)
Drug resistance has unfortunately gone hand-in-hand with HIV-1 drug development. It develops as the consequence of failure to achieve and maintain complete suppression of replication. Although drug resistance is a major cause of treatment failure, several factors contribute to incomplete virus suppression and lead to the selection of drug-resistant viral variants. These factors include incomplete adherence to treatment, individual variation in drug absorption and/or metabolism, disease stage (e.g., baseline virus load and CD4 cell count), and presence of unrecognized drug-resistant virus (e.g., due to transmission of a drug-resistant strain of HIV-1 or previous antiretroviral therapy). The HIV field eventually made use of many barriers to resistance, including pharmacokinetic ones, as demonstrated by achieving much higher levels of PIs by boosting with ritonavir, a weak PI on its own (55). As such, failure of the ritonavir-boosted PI is rarely accompanied by the emergence of PI resistance in patients receiving these drugs for the first time.
Emergence of resistance can also be delayed or prevented by use of certain drug combinations. For example, combined use of lamivudine with zidovudine significantly delays the emergence of zidovudine resistance. Combinations with potent activity that require a broad range of viral mutations for the development of resistance are particularly beneficial. Moreover, the fitness cost of certain drug-resistant mutations reduces the replication capacity of HIV-1 sufficiently so that continued drug administration confers ongoing clinical benefit despite the presence of high-level drug resistance. The adoption of genotypic and phenotypic resistance testing as standard of care has greatly improved selection of appropriate antiretroviral regimens.
When should therapy be initiated? HIV, HBV, HCV
(Dr Robert Schooley, University of California, San Diego)
Despite the existence of consensus guidelines for each viral infection, making the decision to initiate therapy requires that therapeutic goals, as well as the risks and benefits of therapeutic interventions, be decided in the context of each patient at each point in time. Furthermore, decisions about starting therapy related to these three particular viral infections are highly interrelated.
For HIV, the goal of therapy is to increase disease-free survival. The risk for HIV-associated morbidity and mortality increases with rising plasma HIV-1 RNA level and falling CD4 cell count. Although the considerations related to HIV-1-associated opportunistic infections and malignancies were previously paramount, it is now recognized that uncontrolled HIV replication is associated with morbidity and mortality from other coexisting conditions, including renal disease, cardiovascular disease, and hepatitis B and C infections. With better tolerated, coformulated antiretroviral agents, antiretroviral therapy is indicated for patients with symptomatic disease at any CD4 cell count and should be considered for those who are asymptomatic with CD4 cell counts <350 CD4 cells/mm3 (56). Therapy should also be considered for HIV-1-infected persons with coinfection with HBV or HCV, HIV-1-associated nephropathy, or who are at substantial risk for cardiovascular disease at any CD4 cell count (57).
HBV is treated to prevent ongoing liver damage and the development of hepatocellular carcinoma. As therapy is rarely curative and long-term efficacy is limited by drug resistance, patient age, severity of liver disease, and likelihood of response must be balanced against the potential complications of therapy. In general, those with higher plasma levels of HBV DNA and/or histological evidence of liver inflammation are more likely to achieve clinical benefit from therapy (58).
In contrast to HIV and HBV, HCV treatment aims to eliminate the virus. If the virus is eliminated, HCV-related morbidity and mortality are reduced. Criteria for recommending therapy for HCV infection reflect a balance of considerations related to the likelihood of HCV-associated morbidity and mortality, the likelihood of achieving treatment goals and the short- and long-term risks of HCV therapy (59,60). Current indications for therapy will likely change significantly with the availability of small molecular inhibitors.
UNIQUE POPULATIONS: HIV/HBV, HIV/HCV
HIV/hepatitis B coinfection
(Dr Douglas Dieterich, Mount Sinai School of Medicine)
As a result of shared modes of transmission, ∼10% of HIV-infected persons have chronic hepatitis B infection (61). HIV infection is associated with increased risk of chronicity after acute infection, decreased hepatitis B e antigen (HBeAg) loss, increased HBV DNA levels, accelerated liver disease progression, and increased liver mortality. The clinical management of HBV-coinfected patients is complicated by the fact that several antiviral agents inhibit HIV-1 reverse transcriptase, as well as the HBV polymerase, including several that have been approved in the United States for the treatment of HIV: lamivudine (1995), emtricitabine (2003), tenofovir (2001), as well as the fixed-dose combination of tenofovir-emtricitabine (2004) (10). In addition, two medications approved for HBV treatment may also have anti-HIV activity: adefovir (anti-HIV activity at higher doses, 2002) and entecavir (2005) (62,63). Serious hepatic complications have been observed in HIV/HBV-coinfected patients after the discontinuation of antiretroviral therapy that was also acting to suppress HBV replication, leading to enhanced safety warnings for these agents (64,65). Further, the use of entecavir to treat hepatitis B in HIV-infected persons not receiving antiretroviral therapy has been associated with the selection of a mutation in the HIV reverse transcriptase (M184V), which confers resistance to lamivudine and emtricitabine (62,66).
Although there are limited data on the optimal management of HBV in HIV-infected persons, the principles of dual activity and prevention of viral resistance are the basis for treatment recommendations (10,58,67,68). Current guidelines for the treatment of HIV infection indicate that persons with HBV coinfection should be considered on the basis of which disease (HIV and/or HBV) requires antiviral treatment. When HIV alone requires treatment, fully suppressive antiretroviral therapy, which includes the combination of tenofovir and emtricitabine or lamivudine, is recommended, which is also the case if both infections require treatment. If treatment is needed for HBV but not for HIV, fully suppressive antiretroviral therapy, which includes the combination of tenofovir and emtricitabine or lamivudine, should also be considered. If HIV treatment is not an option, peginterferon alfa or adefovir alone or in combination with telbivudine may be considered.
Although effective in suppressing HBV replication in many patients with HIV, current treatment approaches have been associated with relatively low likelihood of HBsAg and HBeAg seroconversion compared with HBV-infected patients without HIV (69). To avoid the development of HBV-resistant mutants with long-term therapy, the DHHS Panel on Antiretroviral Guidelines for Adults and Adolescents has endorsed the routine use of combination anti-HBV therapy (10). However, there are few data from controlled trials to support this approach.
Novel approaches to treatment are needed in HIV-HBV coinfection. Given the shared features of HBV and HIV, anti-HBV drugs that target the polymerase should be thoroughly tested in vitro and in vivo to understand the impact, if any, on HIV. The efficacy and safety of immunological agents (e.g., IFN alfa) may also be altered in the setting of HIV disease (69,70). Studies of novel and existing HBV therapies must be designed and conducted in HIV-infected persons rather than adapted from mono-infected populations.
Therapeutic considerations in HIV-HCV coinfection
(Mark Sulkowski, Johns Hopkins University)
Because of the high prevalence of HCV and more rapid progression of its liver disease in the setting of coinfection with HIV, HCV cirrhosis is now a leading cause of death among persons with HIV. Current guidelines recommend that all HIV-infected persons with acute or chronic HCV infection should be considered for HCV treatment (59,67,71). On the basis of randomized, controlled trials, PegIFN/RBV is the recommended treatment for hepatitis C in HIV-infected persons. Sustained virological response (SVR) rates range from 14% to 29% for HCV genotype 1 infection and 43-73% for HCV genotypes 2 and 3 infection (72,73,74,75). Clinical development of direct-acting antiviral agents for hepatitis C in HIV-infected patients is challenging because of the high likelihood of drug interactions, increased side effects, and concern regarding treatment efficacy and compliance. Nevertheless, in light of the important medical need, the U.S. Food and Drug Administration (FDA) Antiviral Products Advisory Committee emphasized the importance of testing new anti-HCV drugs in this patient group before the initial approval of the drug (76).
Overall, SVR rates observed in HIV-coinfected patients are lower than those reported in patients with HCV monoinfection. High HCV RNA in coinfected patients likely contributes to this. The effect of HCV RNA level is most pronounced in patients with genotype 1 infection in whom the SVR rate was 18% and 62% with high (>800,000IU/ml) and low HCV RNA levels, respectively (74,77). By comparison, SVR rates in HIV seronegative, genotype 1-infected persons treated with the same PegIFN/RBV regimen were 36% and 55% with high and low HCV RNA levels, respectively (78). Other factors influencing HCV treatment include CD4 cell count and concurrent HIV medication. Specifically, several nucleoside analogs increase the toxicity of PegIFN/RBV (zidovudine ⇒ anemia; didanosine ⇒ mitochondrial toxicity) or decrease viral response (abacavir) (79,80,81). Well-controlled HIV disease, genotype 2 or 3, and genotype 1/low HCV load are associated with higher SVR among coinfected patients (82).
The potential use of DAA agents against hepatitis C in coinfected patients raises a number of important considerations: (1) drug-drug interactions with antiretroviral drugs. For example, some HIV PIs inhibit and/or induce cytochrome P450 enzymes, which may increase or decrease levels of agents with similar metabolic pathways (83). Further, many NRTIs require phosphorylation through intracellular kinases to active triphosphate forms, and anti-HCV nucleoside analog polymerase inhibitors may compete with them for phosphorylation (84); (2) HCV drug resistance due to insufficient antiviral activity of PegIFN/RBV; (3) medication toxicity/intolerability (e.g., anemia, rash); and (4) adherence to complex medical regimens targeting both viruses. Despite these challenges, the high prevalence of HCV disease and decreased efficacy of current treatment paradigms underscore the importance of developing innovative approaches to HCV treatment in coinfected patients.
THE CURRENT TREATMENT LANDSCAPE: HEPATITIS B AND HEPATITIS C
FDA: priorities and obstacles in HBV, HCV drug development
(Russell D. Fleischer, Unites States Food and Drug Administration)
Development of therapeutics targeting HBV and HCV is fraught with challenges related to differences between clinical and regulatory needs for approval. Although clinical research is mainly focused on defining whom to treat, when to initiate therapy, how to monitor treatment response, and when to change or stop therapy, FDA has three specific concerns: (i) dosing, (ii) safety, and (iii) efficacy.
Currently, all drugs approved for treatment of HBV target a single site of viral replication (58). Most require a long duration of use to achieve clinically important end points, which is complicated by the emergence of viral resistance. Because of these limitations, the FDA has identified the advancement of promising compounds as a priority in the field of HBV therapeutics. Efforts should be focused on new drugs that increase the rate of viral suppression, decrease the rate of resistance, and decrease the duration of therapy. Clinical trials evaluating new drugs targeting HBV must include both e-Ag-negative and e-Ag-positive patients and include both treatment-naive and treatment-experienced patients.
As there are a greater number of compounds in development that target chronic HCV than HBV, priority in the field of HCV therapeutics lies in identifying those drugs that increase rates of SVR and improve the tolerability of therapy. Opportunities for drug development exist for three different HCV populations: (i) treatment-naive patients with genotype 1, histological injury, and a high baseline viral load; (ii) genotype 1 nonresponders to earlier therapy; and (iii) compensated cirrhotics. In addition, new therapeutics can be studied as a single agent, a replacement for ribavirin, or in combination with current standard of care (pegylated-IFN plus ribavirin) that allows for a shorter duration of treatment or increases efficacy.
For both HBV and HCV, there are certain populations that pose additional challenges to developing safe and effective treatments. These include the pediatric population, those born to mothers with chronic infection, patients with HIV coinfection, and decompensated pre- and posttransplant cirrhotics (85). Given the chronicity of disease in children and the severity of disease in coinfection and in cirrhotics, these populations deserve particular attention when developing new therapeutics.
What should end points of clinical trials in HBV and HCV be?
(Dr Kenneth Sherman, University of Cincinnati)
Ideally, studies regarding drug efficacy should measure hard outcomes such as fibrosis progression, mortality, and hepatocellular carcinoma development, but these end points are generally too distant for the clinical research setting. Surrogate markers for outcomes-such as viral load, serum alanine aminotransferase (ALT), and serology-have emerged, enabling a more practical approach to studying new and existing therapeutics.
Our current clinical practice regarding the treatment of chronic HBV has been influenced by landmark trials that have used the histological activity index (HAI) as the primary outcome. Other surrogate end points have included serological outcomes (e.g., HBeAg loss, HBsAg loss, HBeAb appearance), markers of hepatic injury (e.g., ALT), and composite scores (e.g., ALT+HBeAg loss, HBV DNA+ALT).
However, changes in test technology that allow finer quantification of these surrogate measures have advanced dramatically over the past two decades. HBV DNA early hybridization assays could only detect virus at a minimum of 106copies/ml, whereas current real-time polymerase chain reaction (PCR) techniques have increased sensitivity to <20IU/ml (86,87). However, the question of whether there is a clinically relevant difference between viral clearance at <20IU/ml vs. <100IU/ml remains. At least for drugs with a low genetic barrier to resistance, undetectability of HBV DNA using highly sensitive assays at early treatment time points is associated with the lowest rates of long-term resistance (88).
For drugs targeting HCV, progression of fibrosis, clinical decompensation of cirrhosis, and death are hard end points, but SVR remains the ultimate goal. Promising surrogate markers, however, now provide earlier clues to long-term drug efficacy. Rapid viral response (RVR) and complete early viral response (cEVR), or undetectable HCV viral load at 4 and 12 weeks, respectively, have been shown to be strong positive predictors of SVR, but it is as yet unclear whether RVR and cEVR themselves can be used as end points for clinical trials (89,90,91,92). Quantitative histomorphometry by digital image analysis has also been used to estimate fibrosis by calculating the proportion of pixels stained by Sirius red, which stains collagen. The same technique can also measure activated stellate cells with an immunostain specific for smooth muscle actin, thus providing a marker of liver injury. Finally, transient elastography, which noninvasively measures liver tissue elasticity by means of sound waves, provides an indirect measure of fibrosis (93). Given the lack of highly effective, well-tolerated drug regimens for HCV, there is an urgent need to validate these novel surrogate markers in the context of existing therapeutics early in the treatment course to determine their correlations with clinically relevant long-term outcomes.
Reasons for accelerating combo drug development: HBV
(Dr Robert Perrillo, Baylor University)
Resistance to nucleoside analogs occurs whenever monotherapy is used to treat hepatitis B, and reports have begun to emerge of multidrug-resistant HBV after sequential monotherapy. Higher rates of drug resistance are more likely in the following situations: (i) use of a low genetic barrier drug whereby a single-nucleotide substitution is sufficient to lead to viral breakthrough, (ii) high pretherapy HBV DNA levels, (iii) failure to adequately suppress HBV DNA during the first 24-48 weeks of treatment, and (iv) previous nucleoside analog therapy. Situations (ii) and (iii) are interrelated and attributable to the poor proofreading of reverse transcriptase during viral replication, whereas (iv) is explained by archived drug-resistant mutants to the first drug that accelerate resistance to the second drug.
Currently, the major advantage for combination therapy resides in its ability to reduce the incidence of or totally prevent drug resistance. Lower rates of lamivudine resistance are consistently demonstrated when it is used in combination with a second drug, irrespective of whether the second agent is pegylated IFN or another nucleoside analog that lacks the same resistance pattern as lamivudine (94,95). Combination studies of pegylated IFN and lamivudine demonstrate an additive effect on viral suppression during treatment, and the use of emtricitabine with adefovir has been shown to be associated with improved viral kinetics when compared with adefovir alone (95,96). Further studies are needed to confirm these findings using more potent nucleosides with low-resistance rates such as entecavir and tenofovir.
As combination therapy is the cornerstone of successful management of HIV, many infectious disease experts have advocated it as the best initial treatment strategy for hepatitis B. Enthusiasm for this approach to treatment has been declining, however. Combination therapy for previously untreated patients has been met with particular reluctance among hepatologists for several reasons. First, the pivotal clinical trials with nucleoside analogs have not used this strategy, and the available data do not support additional therapeutic efficacy when compared with monotherapy. Second, as mentioned above, very potent nucleoside analogs with extremely low long-term resistance rates are now available. Third, many patients with HBV have limited access to antiviral therapy because of its relatively high cost. Additional costs of combination therapy could be offset by combining two nucleoside analogs into a single tablet at an affordable price, but industry-sponsored antiviral research and drug development has not moved in this direction. From a mechanistic standpoint, it needs to be remembered that none of these drugs have immunoregulatory effects, and they work on the same pathways in the viral replication cycle. Competition for host-derived thymidine kinase necessary for drug activation might explain the lack of additive efficacy.
Taken together, the above observations invite further studies in which pegylated IFN is used in combination with a potent, low-resistant nucleoside analog and compared with monotherapy with the same nucleoside analog. It may be useful to examine viral kinetic end points during the early phase of treatment in these studies, and rates of HBsAg clearance would also be worth examining because of the accelerated clearance of this marker with IFN.
The above concerns should not be interpreted to mean that there is never an advantage in using combination nucleoside analog therapy in treatment-naive patients. Individuals with relatively low HBV DNA levels have a low risk of drug failure or resistance, and so are unlikely to need initial combination therapy. The opposite is likely to be true for individuals with longstanding high viral replication, because in this case the inherent mutability of HBV over a prolonged interval would allow for a vast quasispecies diversity including HBV mutants with reduced susceptibility to one or more nucleoside analogs. One way of identifying the highest risk groups, therefore, would be to stratify treatment in future clinical trials according to HBV DNA level, duration of infection (when known), previous antiviral therapy, or any other factors felt to be associated with increased likelihood of drug resistance. A further study of the baseline features of patients who developed resistance during recent phase 3 trials could also help to identify risk factors for resistance. The results of these investigations can then provide a stronger latticework for the selective and appropriate use of combination therapy in the future.
Accelerating the development of combination therapy for HCV
(Dr Ira Jacobson, Weill Cornell Medical College)
Since the introduction of the two PegIFNs, we have been immersed in an "era of refinement" during which issues such as optimal dosing, duration of therapy, challenging populations, and strategies for nonresponders have been studied extensively. We now stand poised at the threshold of a new era in HCV therapy, with recent data showing improved rates of SVR when a PI is added to the standard PegIFN/RBV regimen, possibly even with shortened duration of therapy. As monotherapy, DAA agents of several classes have been shown to induce significant viral suppression, with the addition of PegIFN/RBV or even PegIFN alone conferring additional viral suppression and/or markedly inhibiting the development of resistance to the direct-acting antiviral agent (50). In the PROVE1 and PROVE2 studies of genotype 1 treatment-naive patients treated with the PI telaprevir, viral breakthrough was an infrequent event, especially in patients who experienced RVR (97,98). Similarly, in the SPRINT-1 boceprevir trial in treatment-naive patients, only a small number of patients discontinued for viral breakthrough (99).
These observations, together with in vitro experiments demonstrating the capacity of two or three drug combinations to induce additive or synergistic viral suppression (44), as well as the history of HIV therapy, provide a compelling case for the exploration of combinations of direct-acting antiviral agents. Such combinations will doubtless be studied with IFN as a cornerstone, but ultimately the development of IFN-free regimens is a major goal. An experiment in the chimp model of HCV infection has shown that an IFN-free combination of an HCV protease and polymerase inhibitor can result in SVR (100), providing a tantalizing "proof of concept." Very recently, the INFORM-1 study, an intriguing dose-ranging, exploratory study of R7227, a PI, combined with R7128, a nucleoside polymerase inhibitor, for up to 2 weeks, demonstrated marked viral suppression (101). No viral breakthroughs were reported, although sequencing data are pending at the time of writing. Data from the RBV-free arm of PROVE2 (98), the RBV-free arm of PROVE3, a study on telaprevir in earlier nonresponders (102), and the low dose RBV arm of SPRINT-1 (99) have provided a compelling case for the role of RBV in preventing the emergence of resistant variants. Such data provide a foundation for the early exploration of IFN-free regimens consisting of two DAA agents plus RBV.
Given the universal interest in studying combinations of DAA agents, the timeline for the development of such combinations is a critical issue. Patients, health-care providers, the pharmaceutical industry, and regulatory agencies are all major stakeholders in this process. Patients and physicians should test combinations with promising potential efficacy at the earliest point that there are preclinical drug interaction, safety, and efficacy studies. Industry must understandably be cautious about the potential for drug interactions or unexpected adverse events that may occur with the use of drugs in combination studies, balancing these considerations with the prospect of taking what would be a momentous leap forward in the field (not to mention the need to remain competitive as others move ahead). Of course, the FDA and other regulatory agencies must put safety as the paramount consideration in evaluating each drug of a proposed regimen before they can be combined. Avoidance of resistance is also a regulatory concern, underscoring the importance of careful observations, including sensitive sequencing studies, in early studies of combination therapy. The pilot study combining a protease and polymerase inhibitor cited above (101) was performed in Australia and New Zealand. It is hoped that such studies will be regarded as feasible in the United States and Europe on the basis of data on each potential component of potential combination regimens gleaned from pre-phase 3 trials. There is a published indication from the FDA Antiviral Products Advisory Committee that this is in accord with the FDA's perspective to a substantial extent (76). Open communication between industry, regulatory agencies, clinical investigators, and possibly in a manner reminiscent of the history of HIV therapy, patient advocacy groups should prove helpful in moving the field in this direction.
Imagining the future: how therapies and resistance will evolve
(Dr John McHutchison, Duke University)
The development of newer therapies for the treatment of patients with chronic HCV infection (Figure 6) continues to present major challenges with respect to efficacy, duration of therapy, safety, and tolerability. Although the ideal drugs would attain >80% response rates with minimal relapse and maximum tolerability, current drug regimens targeting HCV genotype 1 achieve a 40% rate of SVR, with over 20% relapse, 25-30% nonresponse, and 5-15% discontinuation rates secondary to adverse effects. In an attempt to improve on the current standard regimen of PegIFN/RBV, trials evaluating new therapies are increasing in complexity and have explored the addition of a third agent for the same or shorter duration of treatment, using a "lead-in" phase with either the standard therapy followed by the new agent or vice versa, or replacing either IFN or RBV with the new agent (44). Some of these strategies have been successful, whereas others have not.
Hepatitis C drug development: 2009 (courtesy of Dr John McHutchison). IFN, interferon
One of the major future issues confronting the treatment of HCV with direct-acting antivirals will be drug resistance. Currently, there are no standards regarding the evaluation of HCV resistance-whether it be a standard nomenclature for resistant variants, definitions of virological breakthrough, guidelines for when and how often to test for resistance during therapy, or prediction models for how the HCV virus will react given a certain patient, genotype, viral load, and combination of drugs, akin to advances seen in HIV research. The FDA has provided a helpful framework for conducting and submitting HCV resistance data (103). As new therapies targeting HCV emerge, limiting or curtailing resistance will become critical, and as important an end point as efficacy, safety, and tolerability. Whatever future HCV triple or even quadruple treatment regimens may look like, the beginning of this era looks bright in terms of achieving higher sustained response rates and curtailing the duration of therapy for our HCV-infected patients.
|
|
| |
| |
|
|
|