| |
Efficacy of boceprevir, an NS3 protease inhibitor, in combination with peginterferon alfa-2b and ribavirin in treatment-naive patients with genotype 1 hepatitis C infection (SPRINT-1): an open-label, randomised, multicentre phase 2 trial - in the Lancet Aug 9 Epub -
|
| |
| |
publication pdf attached, figure 5 is very useful
"Rapid virological response was highly associated with SVR in all treatment groups. In the 28-week treatment groups, 82% (54/66) of patients in the PR4/PRB24 group and 74% (32/43) in the PRB28 group who had rapid virological response achieved SVR. In the 48-week treatment groups, 94% (62/66) of patients assigned to PR4/PRB44 and 84% (32/38) assigned to PRB48 who achieved undetectable hepatitis C virus RNA by week 4 of boceprevir achieved SVR. Achievement of undetectable hepatitis C virus RNA between weeks 4 and 12 of boceprevir therapy was also highly predictive of SVR in the 48-week treatment groups (table 3; figure 4). We noted a greater SVR in patients who cleared virus between weeks 4 and 12 of boceprevir in the PR4/PRB44 group compared with the PR4/PRB24 group (table 3).....
Thus, in the treatment of genotype 1 hepatitis C virus, nearly two-thirds of patients achieved undetectable hepatitis C virus RNA levels at week 4 of boceprevir therapy after PR4, and these individuals can be treated for 28 weeks with high SVR. Of the additional 18% (19/103) of patients who go on to achieve undetectable hepatitis C virus RNA between weeks 4 and 12 of boceprevir therapy, 79% (15/19) of patients benefit from extending therapy with peginterferon alfa-2b, ribavirin, and boceprevir to 48 weeks. Only one patient in the boceprevir groups who developed undetectable hepatitis C virus RNA after week 12 of boceprevir therapy went on to SVR."
"The lead-in (PR4) allowed us to examine the relation of peginterferon and ribavirin responsiveness at week 4 to SVR with boceprevir-containing regimens. In the PR4 28-week or 48-week groups, SVR was similar in participants with greater than 1·5 log10 reduction in hepatitis C virus RNA from baseline before the addition of boceprevir. Higher SVR was noted in participants who received PRB for 44 weeks with less than 1·5 log10 reduction from baseline at PR4 (figure 5). In patients with less than 1 log10 reduction with PR4, 55% (95% CI 32-76) SVR was noted in the PR4/PRB44 group.....
The lead-in can identify null responders to peginterferon alfa-2b and ribavirin, who seem to be at greatest risk for treatment failure with specifically targeted therapies and for development of resistance. However, in our cohort, a substantial proportion of null responders during the lead-in period went on to achieve SVR with the addition of boceprevir.....In the novel lead-in approach, we recorded increased SVR and a reduction in relapse and breakthrough, and allowed for potential determination of treatment duration on the basis of responsiveness to the PR4 lead-in. However, in the direct comparison between lead-in and non-lead-in groups, relapse reduction did not differ significantly, although the absence of a statistically conclusive result is not surprising since the sample size did not allow detection of modest differences between lead-in and non-lead-in groups. The mutations recorded in participants with viral breakthrough were consistent what those that have been previously reported with NS3 inhibitors with no new mutations noted.23-26 The clinical relevance of these mutations is unknown, and long-term follow-up is in progress."
"The SVR rate for black people in the PR48 control group was 13% (two of 16) and as high as 53% (eight of 15) in patients treated with boceprevir for 48 weeks (table 4). In patients with cirrhosis, the SVR rate was 67% (ten of 15) in the combined longer duration boceprevir groups versus 25% (two of eight) in the control group; in patients without cirrhosis, the SVR rate for the combined longer duration boceprevir groups was 71% (136/191) compared with 39% (37/96) for the control group."
"We also noted increased rates of SVR in patients who developed anaemia (haemoglobin <100 g/L) irrespective of treatment group. Epoetin alfa was used by 40% (236/595) of patients and was allowed at investigator discretion. The use of this drug in those with anaemia was also associated with an improved SVR (table 4).....
A study suggested that development of anaemia with haemoglobin less than 100 g/L is associated with increased SVR in patients receiving pegylated interferon and ribavirin; anaemia is potentially a surrogate marker of increased ribavirin concentration and the addition of epoetin alfa might allow patients to remain on therapy"
"These results are consistent with those recorded in two trials of another NS3 protease inhibitor, telaprevir, in combination with peginterferon and ribavirin, in populations that excluded those with histological cirrhosis and had fewer black people.17, 18 In one of these studies,17 undertaken in the USA, the SVR rate in the telaprevir group receiving 24 weeks of treatment with 12 weeks of telaprevir and 24 weeks of peginterferon and ribavirin was 20% higher than in the control group of 48 weeks of peginterferon and ribavirin (61% [48/79] vs 41% [31/75]); and in the telaprevir group receiving 48 weeks of treatment with 12 weeks of telaprevir added to 48 weeks of peginterferon and ribavirin, the SVR rate was 26% higher than it was in the 48-week control group (67% [53/79] vs 41% [31/75]). In the other study,18 undertaken in Europe, the SVR rate in the telaprevir group receiving 24 weeks of treatment with 12 weeks of telaprevir and 24 weeks of peginterferon and ribavirin was 23% higher than in the 48-week control group (69% [56/81] vs 46% [38/82]). Although the study designs and populations differed, in our study, SVR rates were 17-19% higher than in the control group in the 28-week boceprevir group, and 29-37% higher than control in the 48-week boceprevir groups."
Efficacy of boceprevir, an NS3 protease inhibitor, in combination with peginterferon alfa-2b and ribavirin in treatment-naive patients with genotype 1 hepatitis C infection (SPRINT-1): an open-label, randomised, multicentre phase 2 trial
The Lancet, Early Online Publication, 9 August 2010
Dr Paul Y Kwo MD a Corresponding AuthorEmail Address, Eric J Lawitz MD b, Jonathan McCone MD c, Prof Eugene R Schiff MD d, Prof John M Vierling MD e, David Pound MD f, Mitchell N Davis DO g, Joseph S Galati MD h, Stuart C Gordon MD i, Natarajan Ravendhran MD j, Prof Lorenzo Rossaro MD k, Frank H Anderson MD l, Prof Ira M Jacobson MD m, Raymond Rubin MD n, Kenneth Koury PhD o, Lisa D Pedicone PhD o, Clifford A Brass MD o, Eirum Chaudhri MD o, Janice K Albrecht PhD o
Summary
Background
Peginterferon plus ribavirin achieves sustained virological response (SVR) in fewer than half of patients with genotype 1 chronic hepatitis C virus infection treated for 48 weeks. We tested the efficacy of boceprevir, an NS3 hepatitis C virus oral protease inhibitor, when added to peginterferon alfa-2b and ribavirin.
Methods
In part 1 of this trial, undertaken in 67 sites in the USA, Canada, and Europe, 520 treatment-naive patients with genotype 1 hepatitis C virus infection were randomly assigned to receive peginterferon alfa-2b 1·5 µg/kg plus ribavirin 800-1400 mg daily for 48 weeks (PR48; n=104); peginterferon alfa-2b and ribavirin daily for 4 weeks, followed by peginterferon alfa-2b, ribavirin, and boceprevir 800 mg three times a day for 24 weeks (PR4/PRB24; n=103) or 44 weeks (PR4/PRB44; n=103); or peginterferon alfa-2b, ribavirin, and boceprevir three times a day for 28 weeks (PRB28; n=107) or 48 weeks (PRB48; n=103). In part 2, 75 patients were randomly assigned to receive either PRB48 (n=16) or low-dose ribavirin (400-1000 mg) plus peginterferon alfa-2b and boceprevir three times a day for 48 weeks (low-dose PRB48; n=59). Randomisation was by computer-generated code, and study personnel and patients were not masked to group assignment. The primary endpoint was SVR 24 weeks after treatment. Analysis was by intention to treat. This study is registered with ClinicalTrials.gov, number NCT00423670.
Findings
Patients in all four boceprevir groups had higher rates of SVR than did the control group
- (58/107 [54%, 95% CI 44-64], p=0·013 for PRB28;
- 58/103 [56%, 44-66], p=0·005 for PR4/PRB24;
- 69/103 [67%, 57-76], p<0·0001 for PRB48; and
- 77/103 [75%, 65-83], p<0·0001 for PR4/PRB44;
vs 39/104 [38%, 28-48] for PR48 control).
Low-dose ribavirin was associated with a high rate of viral breakthrough (16/59 [27%]), and a rate of relapse (six of 27 [22%]) similar to control (12/51 [24%]).
Boceprevir-based groups had higher rates of anaemia (227/416 [55%] vs 35/104 [34%]) and dysgeusia (111/416 [27%] vs nine of 104 [9%]) than did the control group.
Interpretation
In patients with untreated genotype 1 chronic hepatitis C infection, the addition of the direct-acting antiviral agent boceprevir to standard treatment with peginterferon and ribavirin after a 4-week lead-in seems to have the potential to double the sustained response rate compared with that recorded with standard treatment alone.
Funding
Merck.
Introduction
Chronic hepatitis C virus affects about 170 million people worldwide. Cirrhosis induced by hepatitis C virus is the most common indication for liver transplantation and is a major contributor to the worldwide increase in the incidence of hepatocellular cancer.1, 2 Standard-of-care treatment of genotype 1 hepatitis C virus is pegylated interferon and ribavirin for 48 weeks, which results in sustained virological response (SVR) in about 40-50% of individuals.3-5 SVR rates for black patients treated with standard of care are substantially lower; in two studies undertaken almost exclusively in genotype 1 individuals, 19-28% of black people achieved SVR versus 52% of non-Hispanic white people.6, 7 Those who achieve SVR can have long-term benefits with improvement in degrees of liver fibrosis, reduction in complications of chronic liver disease, and improved quality of life.8 Studies have shown that response-guided therapy can allow tailoring of duration of treatment, with week 4 viral clearance (rapid virological response) allowing shorter duration of therapy than for those who clear virus at week 12 (complete early virological response).9-12
Up until now, treatment for this disease has consisted of therapies to stimulate the immune system and interfere in a non-specific manner with viral replication. Research has focussed on therapies that inhibit hepatitis C virus proteins that are essential for intracellular replication; these drugs are referred to as direct-acting antiviral agents.13 Boceprevir is a novel peptidomimetic NS3 protease inhibitor that forms a covalent reversible complex with the NS3 protease in vitro and has shown potent antiviral activity in the hepatitis C virus replicon system, and in patients who previously showed no response to peginterferon administered with or without ribavirin.14, 15 In a dose-ascending study in null responders,16 boceprevir, when given in combination with peginterferon alfa-2b and ribavirin, was associated with a modest incremental haemoglobin reduction, as has been recorded with other direct-acting antiviral agents in the NS3 inhibitor class.17, 18 The NS3 protease inhibitor telaprevir has also shown significantly higher rates of SVR than has standard of care in patients with genotype 1 disease when given for 12 weeks in combination with regimens of peginterferon and ribavirin lasting 12, 24, or 48 weeks.17, 18 Although rates of SVR in the telaprevir groups were higher than were those recorded with standard of care, investigators noted higher drop-out rates due to increased side-effects.
The aim of the hepatitis C virus SPRINT-1 (Serine Protease Inhibitor Therapy-1) study was to establish the safety and efficacy of boceprevir when added to peginterferon and ribavirin. We investigated treatment durations of 28 weeks versus 48 weeks, with and without a 4-week lead-in of peginterferon alfa-2b and ribavirin before the addition of boceprevir for 24 or 44 weeks, and the efficacy of low-dose ribavirin. The rationale for the 4-week lead-in was to allow pegylated interferon and ribavirin to reach steady-state concentrations before the addition of boceprevir such that backbone drug concentrations would be at an optimum and potentially reduce the likelihood for emergence of drug-resistant mutations by reducing viral levels.19-21 Part 2 of this study, which was added after enrolment in part 1 was completed, was undertaken to assess the possibility of use of a lower dose of ribavirin to reduce treatment complications, mainly anaemia. Since boceprevir and other NS3 protease inhibitors have been associated with a modest reduction in haemoglobin, weight-based low-dose (400-1000 mg per day) versus standard-dose (800-1400 mg per day) ribavirin was assessed to establish whether efficacy could be maintained while reducing anaemia. Lastly, we examined the rates of viral clearance and SVR.
Results
765 patients were screened and 595 were enrolled in 43 US, four Canadian, and 20 European Union sites. All efficacy and safety analyses were based on 595 patients who were randomly assigned and received at least one dose of medication. Table 1 shows baseline characteristics, and figures 2 and 3 show patient disposition for all groups.
Table 2 shows virological response. Irrespective of treatment duration or use of PR4 as lead-in, SVR rates in all four boceprevir groups in part 1 were significantly better than were those in the PR48 control group. In the 28-week treatment groups, the SVR was 56% (95% CI 46-66) in the PR4/PRB24 group (p=0·005 vs control) and 54% (44-64) in the PRB28 group (p=0·013 vs control). In the 48-week treatment groups, the SVR was 75% (65-83) in the PR4/PRB44 group (p<0·0001 vs control) compared with 67% (57-76) in the PRB48 group (p<0·0001 vs control). In part 2, the SVR for low-dose PRB48 was 36% (24-49).
We noted significantly lower relapse rates in the 48-week treatment groups compared with PR48 control (PRB48, p=0·0079; PR4/PRB44, p=0·0002; table 2). Although the relapse rates were higher in the 28-week than in the 48-week treatment groups, patients in the 28-week groups who had rapid hepatitis C virus RNA clearance at week 4 of boceprevir had substantially lower relapse rates than did those who did not have rapid viral clearance (p<0·0001; table 2). Low-dose PRB was associated with high relapse rates. Of note, we recorded no viral breakthrough in control groups that did not contain boceprevir (table 2). In the boceprevir groups, the lead-in groups were associated with a modestly lower rate of breakthrough than were the groups with no lead in. Combining across treatment groups, the rate of breakthrough in the boceprevir lead-in groups was 4% (nine of 206) compared with 9% (19/210) in the boceprevir groups with no lead in (p=0·057). By population sequencing, the major mutations (in >25% of samples) were V36M, T54S, and R155K, with less common (in 5% to <25%) mutations including T54A, V55A, R155T, A156S, V158I, and V170A (data not shown). Infrequent mutations (in <5% of samples) included V36A, V36L, and I170T (data not shown).
Rapid virological response was highly associated with SVR in all treatment groups. In the 28-week treatment groups, 82% (54/66) of patients in the PR4/PRB24 group and 74% (32/43) in the PRB28 group who had rapid virological response achieved SVR. In the 48-week treatment groups, 94% (62/66) of patients assigned to PR4/PRB44 and 84% (32/38) assigned to PRB48 who achieved undetectable hepatitis C virus RNA by week 4 of boceprevir achieved SVR. Achievement of undetectable hepatitis C virus RNA between weeks 4 and 12 of boceprevir therapy was also highly predictive of SVR in the 48-week treatment groups (table 3; figure 4). We noted a greater SVR in patients who cleared virus between weeks 4 and 12 of boceprevir in the PR4/PRB44 group compared with the PR4/PRB24 group (table 3).
Table 3 Rates of sustained virological relapse by time to first undetectable hepatitis C virus RNA
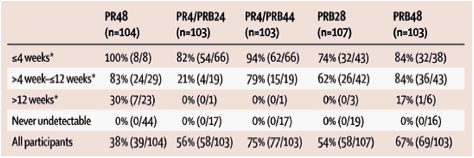
The lead-in (PR4) allowed us to examine the relation of peginterferon and ribavirin responsiveness at week 4 to SVR with boceprevir-containing regimens. In the PR4 28-week or 48-week groups, SVR was similar in participants with greater than 1·5 log10 reduction in hepatitis C virus RNA from baseline before the addition of boceprevir. Higher SVR was noted in participants who received PRB for 44 weeks with less than 1·5 log10 reduction from baseline at PR4 (figure 5). In patients with less than 1 log10 reduction with PR4, 55% (95% CI 32-76) SVR was noted in the PR4/PRB44 group.
A multivariate regression analysis of pooled boceprevir groups in part 1 of the study was done by investigation of baseline factors associated with SVR. These factors included low viral load (�600 000 IU/mL), non-black race, lower platelet count, and genotype 1b. In the control group of this study which received the standard of care, black people and those with cirrhosis had a lower SVR than did participants of non-black race and with no cirrhosis (table 4). The SVR rate for black people in the PR48 control group was 13% (two of 16) and as high as 53% (eight of 15) in patients treated with boceprevir for 48 weeks (table 4). In patients with cirrhosis, the SVR rate was 67% (ten of 15) in the combined longer duration boceprevir groups versus 25% (two of eight) in the control group; in patients without cirrhosis, the SVR rate for the combined longer duration boceprevir groups was 71% (136/191) compared with 39% (37/96) for the control group.
We also noted increased rates of SVR in patients who developed anaemia (haemoglobin <100 g/L) irrespective of treatment group. Epoetin alfa was used by 40% (236/595) of patients and was allowed at investigator discretion. The use of this drug in those with anaemia was also associated with an improved SVR (table 4).
The most common adverse events in the boceprevir groups, as reported by investigators, were fatigue, anaemia, nausea, and headache-a side-effect profile generally similar to that recorded in patients receiving PR48 control. The rate of dysgeusia and anaemia was higher in the boceprevir groups than in other groups (table 5). In the boceprevir groups, we detected nadir haemoglobin concentrations of 85-100 g/L in those who developed anaemia; and haemoglobin concentrations less than 85 g/L were rare (table 6). Dose modifications of ribavirin were similar in boceprevir groups compared with the PR48 control group, and boceprevir dose modifications were rare (table 7). We recorded an overall higher discontinuation rate in the boceprevir groups than in the control group (table 7). Treatment discontinuations attributable to adverse events ranged from 9% to 19% in the groups receiving boceprevir therapy compared with 8% for the PR48 group, with two patients discontinuing treatment because of anaemia in the boceprevir groups (table 7). The rate of adverse events categorised as skin and subcutaneous tissue disorders was similar in boceprevir-containing regimens (159/416, 38%) and the control group (38/104, 37%).
Discussion
The results of this phase 2 trial have shown that an optimum dose of boceprevir (800 mg three times a day), when added to the standard of care for treatment of chronic genotype 1 hepatitis C virus, significantly increased SVR in both 28-week and 48-week regimens compared with the control of peginterferon alfa-2b and ribavirin. Responses in the 48-week boceprevir groups were substantially higher than were those in the 28-week groups, with a near doubling of SVR in the PR4/PRB44 group. The use of low-dose ribavirin in combination with peginterferon and boceprevir, while reducing haematological toxic effects, did not improve SVR rates compared with control.
These results are consistent with those recorded in two trials of another NS3 protease inhibitor, telaprevir, in combination with peginterferon and ribavirin, in populations that excluded those with histological cirrhosis and had fewer black people.17, 18 In one of these studies,17 undertaken in the USA, the SVR rate in the telaprevir group receiving 24 weeks of treatment with 12 weeks of telaprevir and 24 weeks of peginterferon and ribavirin was 20% higher than in the control group of 48 weeks of peginterferon and ribavirin (61% [48/79] vs 41% [31/75]); and in the telaprevir group receiving 48 weeks of treatment with 12 weeks of telaprevir added to 48 weeks of peginterferon and ribavirin, the SVR rate was 26% higher than it was in the 48-week control group (67% [53/79] vs 41% [31/75]). In the other study,18 undertaken in Europe, the SVR rate in the telaprevir group receiving 24 weeks of treatment with 12 weeks of telaprevir and 24 weeks of peginterferon and ribavirin was 23% higher than in the 48-week control group (69% [56/81] vs 46% [38/82]). Although the study designs and populations differed, in our study, SVR rates were 17-19% higher than in the control group in the 28-week boceprevir group, and 29-37% higher than control in the 48-week boceprevir groups.
In black participants and in those with cirrhosis, the addition of boceprevir to standard of care improved SVR. These preliminary results in a fairly small number of patients suggest that the addition of boceprevir to peginterferon alfa-2b and ribavirin will improve SVR in these difficult-to-treat populations.
This study assessed the use of a PR4 lead-in before the addition of boceprevir, as well as the effect of starting all three drugs concomitantly, and compared these groups with PR48 control. In the novel lead-in approach, we recorded increased SVR and a reduction in relapse and breakthrough, and allowed for potential determination of treatment duration on the basis of responsiveness to the PR4 lead-in. However, in the direct comparison between lead-in and non-lead-in groups, relapse reduction did not differ significantly, although the absence of a statistically conclusive result is not surprising since the sample size did not allow detection of modest differences between lead-in and non-lead-in groups. The mutations recorded in participants with viral breakthrough were consistent what those that have been previously reported with NS3 inhibitors with no new mutations noted.23-26 The clinical relevance of these mutations is unknown, and long-term follow-up is in progress.
The viral response during the lead-in could help to predict best possible treatment duration. Patients achieving less than 1·5 log10 reduction in viral level after PR4 benefit most from a treatment duration of 48 weeks, whereas those with greater than 1·5 log10 reduction show similar SVR irrespective of treatment duration of 28 weeks or 48 weeks. The lead-in can identify null responders to peginterferon alfa-2b and ribavirin, who seem to be at greatest risk for treatment failure with specifically targeted therapies and for development of resistance. However, in our cohort, a substantial proportion of null responders during the lead-in period went on to achieve SVR with the addition of boceprevir.16, 27
In all groups, rapid virological response was highly predictive of SVR. We also recorded high rates of rapid virological response in the 48-week treatment groups. Participants who cleared virus between weeks 4 and 12 of boceprevir therapy were more likely to go on to SVR if they received 48 weeks of treatment rather than 28 weeks. Thus, in the treatment of genotype 1 hepatitis C virus, nearly two-thirds of patients achieved undetectable hepatitis C virus RNA levels at week 4 of boceprevir therapy after PR4, and these individuals can be treated for 28 weeks with high SVR. Of the additional 18% (19/103) of patients who go on to achieve undetectable hepatitis C virus RNA between weeks 4 and 12 of boceprevir therapy, 79% (15/19) of patients benefit from extending therapy with peginterferon alfa-2b, ribavirin, and boceprevir to 48 weeks. Only one patient in the boceprevir groups who developed undetectable hepatitis C virus RNA after week 12 of boceprevir therapy went on to SVR.
We noted no new adverse events or treatment-limiting toxic effects associated with boceprevir-containing regimens in this trial compared with those recorded in patients receiving peginterferon and ribavirin. No increases in skin or subcutaneous adverse events were noted in the boceprevir-containing groups compared with the control groups. Higher rates of both anaemia and dysgeusia were noted in the boceprevir-containing regimens than in the control group, although stopping treatment for anaemia was rare. Haemoglobin reductions in the PRB48 low-dose group were less than those in the control group and in any of the full-dose PRB groups in part 1, and similar to the haemoglobin reduction in the PR48 group of part 1 of the study. In this study, use of epoetin alfa was allowed at the investigator's discretion and was associated with improved SVR. A study suggested that development of anaemia with haemoglobin less than 100 g/L is associated with increased SVR in patients receiving pegylated interferon and ribavirin; anaemia is potentially a surrogate marker of increased ribavirin concentration and the addition of epoetin alfa might allow patients to remain on therapy.28 In our study, the development of anaemia and the use of epoetin alfa were associated with improved SVR in the boceprevir-containing regimens. However, since there was no randomisation for use of epoetin alfa in this study, the contributions of anaemia and epoetin alfa use to improved SVR with boceprevir remains to be established. The role of epoetin alfa as an adjuvant in patients receiving pegylated interferon and ribavirin in addition to therapy with direct-acting antiviral agents deserves further study. We recorded a higher drop-out rate in the boceprevir-containing groups than in the control group, as has been noted when other direct-acting antiviral agents are added to peginterferon alfa-2b and ribavirin.17 This findings suggests that treating physicians might need experience with these agents to ensure patient adherence and manage side-effects, as was noted with the introduction of ribavirin to interferon therapy for hepatitis C virus infection.17, 18
There are potential limitations of this study which deserve mention. This was an open-label study with regard to the administration of boceprevir because of the complex study design with comparisons of lead-in and non-lead-in groups, and differing treatment durations. However, all assays were done by an independent commercial laboratory that did not have access to participant treatment assignments. The primary and other key study endpoints were based on hepatitis C virus RNA level-an outcome that is not subject to bias. Another possible limitation concerns the stratification of patients as with or without cirrhosis. We required a liver biopsy sample to be taken within 5 years of enrolment into the study. A patient who tested negative for cirrhosis 5 years before the beginning of the study could have developed cirrhosis in the intervening years. Thus patients with cirrhosis could have been mischaracterised as being non-cirrhotic, biasing the results in favour of the population with this disease. Therefore, the promising results obtained in patients with cirrhosis who received boceprevir will need confirmation in larger trials that are in progress.
In conclusion, boceprevir, in combination with pegylated interferon and ribavirin, achieved high SVR rates with 28 weeks of therapy in most patients and is safe and effective for use up to 48 weeks in the few patients who benefit from longer duration of therapy. We also recorded increased response rates in difficult-to-treat groups, including black participants and those with cirrhosis. The use of PR4 lead-in before the addition of boceprevir improves SVR over a 48-week duration, and reduces viral breakthrough and relapse. A large confirmatory trial is in progress and will define the best treatment regimen for the use of boceprevir in the treatment of genotype 1 chronic hepatitis C virus infection.
|
|
| |
| |
|
|
|