 |
|
|
| |
Clinical Pharmacology at CROI 2011 (HIV & HCV)
|
| |
| |
Courtney V. Fletcher, Pharm.D.
Dean and Professor
College of Pharmacy
University of Nebraska Medical Center
986000 Nebraska Medical Center
Omaha, NE 68198
The 18th Conference on Retroviruses and Opportunistic Infections (CROI) was held in Boston, MA from February 28 to March 3, 2011. CROI continues to be the premier HIV-focused scientific meeting. In this report I will highlight abstracts focused on pharmacologic issues I think are of broad interest or might benefit from some expert clarification. I will discuss abstracts in four broad categories: advances in therapeutics, drug interactions, compartmental penetration and adverse reactions.
Abbreviations
%CV, percent coefficient of variation
ABC, abacavir
ACTG, adult AIDS clinical trials group
APV, amprenavir
ARV, antiretroviral drug
ART, antiretroviral drug therapy
AUC, area under the concentration-time curve
ATV, atazanavir
BOC, boceprevir
Cmin, minimum drug concentration
CNS, central nervous system
CSF, cerebrospinal fluid
CYP, cytochrome P450 drug metabolizing enzymes
DRV, darunavir
ddI, didanosine
EFV, efavirenz
EVG, elvitegravir
FTC, emtricitabine
ETR, etravirine
fAPV, fosamprenavir
IC50, concentration of drug required to inhibit viral replication in vitro by 50%
IDV, indinavir
IM, intramuscular
IQ, inhibitory quotient
3TC, lamivudine
LPV, lopinavir
MVC, maraviroc
NVP, nevirapine
NRTI, nucleoside reverse transcriptase inhibitor
NNRTI, non-nucleoside reverse transcriptase inhibitor
PACTG, pediatric AIDS clinical trials group
PBMCs, peripheral blood mononuclear cells
PD, pharmacodynamic
PG, pharmacogenetics/pharmacogenomics
PK, pharmacokinetic
PI, inhibitor of HIV protease
PrEP, pre-exposure prophylaxis
r or RTV, ritonavir
RAL, raltegravir
RBT, rifabutin
RBV, ribavirin
SQV, saquinavir
SC, subcutaneous
TDF, tenofovir disoproxil fumarate
TFV, tenofovir
TVR, telaprevir
TDM, therapeutic drug monitoring
TPV, tipranavir
ZDV, zidovudine
I. Advances in Therapeutics: Pre-Exposure Prophylaxis (PrEP) and Treatment of Hepatitis C Virus (HCV) Infection.
I think the 18th CROI will be recognized for two major therapeutic advances - PrEP and new drug to treat HCV infection. In this section below I will highlight these therapeutic advances as well as comment on additional clinical pharmacology issues relevant to these topics, such as new formulations, compartmental penetration and drug-drug interactions.
PrEP
iPrEx: FTC/TDF is safe, well tolerated and reduces acquisition of HIV-infection in MSM by 44%; the reduction in risk was 92% for those with detectable drug.
Robert Grant (Abstract 92) provided an updated analysis of the iPrEx study since publication on November 23, 2010. Overall, this study of oral FTC/TDF found a 42% reduction in HIV infection compared with placebo in the updated analysis. FTC/TDF was safe and well tolerated. There was a small but significant decrease in bone mineral density associated with FTC/TDF (Abstract 94LB).
Peter Anderson (Abstract 96LB) led the iPrEx pharmacology effort to measure plasma and intracellular concentrations of FTC and tenofovir (TFV). This effort provided insight into adherence, safety, resistance and the protection from HIV-infection that otherwise would not have been elucidated. For example, all infections in the active arm of the primary analysis were associated with undetectable (91%) or low (9%) drug concentrations. When the presence of detectable drug was then considered, the reduction in the risk of HIV-infection was 92%. These data indicate that a significant reduction in the overall efficacy of oral FTC/TDF for PrEP is a result of adherence, and highlight the need for strategies to achieve high levels of adherence in future studies and for an eventual rollout for community use.
Rectal application of a 1% TFV vaginal gel had suboptimal tolerance and acceptance.
Abstract 34LB described a phase I study of rectal application of the 1% TFV vaginal gel, which has been shown to reduce the risk of HIV-infection in women (CAPRISA 004). 18 healthy volunteers received a single oral dose of TDF (300mg) and then were randomized to a single rectal dose of the gel or placebo and 7 days of rectal dosing. As expected, plasma TFV concentrations after oral dosing were much higher, 30-fold for AUC, than rectal. The plasma AUC was higher after 7 days of rectal dosing than after the single dose. TFV-diphosphate concentrations were detectable in rectal tissue and were greater after 7 days of rectal application that after the single application. Only the 7-day rectal application strategy inhibited HIV-infectivity in ex vivo biopsies, suggesting single use administration may not be effective. Rectal application was not completely safe and poorly accepted by the volunteers. The suboptimal tolerance, particularly the lower gastrointestinal tract (GI) side effects, may arise in part from the hyperosmolar nature of the vaginal gel product.
A reformulated TFV gel has improved safety.
The poor tolerance of rectal application of the 1% TFV vaginal gel may be a result of it's hyperosmolar characteristic. Abstract 983 described the reformulation of the TFV gel that achieved a 74% reduction in osmolality. There was no change in the anti-HIV activity of the reformulated gel compared with the original with IC50 values of 2.4 μM and 2.9 μM, respectively, and ≥ 3 log reductions of HIV p24 in cell culture experiments with both formulations. This new formulation appears to warrant human study to see if these improved characteristics overcome problems found with the phase I study of rectal administration of the 1% vaginal gel described above.
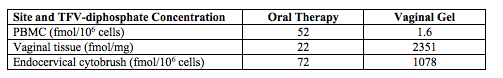
A vaginal gel formulation achieves higher intracellular TFV concentrations in vaginal tissue than oral TDF.
144 women not infected with HIV in the US and Africa participated in this study where they received 300 mg of oral TDF, a 40 mg vaginal gel, or both products daily for 6 months (Abstract 35LB). The data show oral therapy achieves higher concentrations of TFV-diphosphate (the pharmacologically-active component) in peripheral blood mononuclear cells, while the vaginal gel achieves higher concentrations in vaginal tissue.
Single dose intracellular concentrations of FTC and TFV are lower than after repetitive dosing.
Peter Anderson and colleagues measured the intracellular concentrations of FTC and TFV over 24 hours following a single dose of FTC/TDF (200/300mg) in healthy volunteers. The peak intracellular concentration of FTC occurred at 4 hours after the dose, while the peak for TFV occurred at 24 hours. The FTC intracellular concentrations in PBMC were about 30% of what would be expected at steady state, and those of TFV approximately 15% of steady state (Abstract 641).
The results of abstracts 35LB and 641 have important implications for designing and predicting the efficacy of future PrEP studies. Abstract 35LB highlights the differences in concentrations achieved depending on the method of dosing: TFV-diphosphate concentrations were 100-fold higher when TFV was administered with a vaginal gel compared with oral administration. There is interest in episodic dosing regimens for PrEP. Abstract 641 shows that following oral dosing peak intracellular levels of FTC are reached in 4 hours, but peak concentrations of TFV do not occur until 24 hours post dose. In her plenary address at CROI, Connie Celum noted the requirements of PrEP are to deliver the right drug profile, in the right place at the right time. These abstracts provide fundamental pharmacokinetic information necessary for designing PrEP strategies to do so.
Hepatitis C
Clinical trials of telaprevir (TVR) and boceprevir (BOC) each combined with pegylated interferon (IFN) and ribavirin (RBV) for treatment of hepatitis C infection (HCV) showed sustained virologic response could be significantly improved over that achieved with only IFN and RBV in patients who were not co-infected with HIV (Abstracts 115, 116; plenary address of Stefan Zeuzem). These HCV protease inhibitors appear to be much needed new drugs. It has been of concern to the HIV community that these investigational agents have advanced to such a late stage of development without substantive work in HIV-HCV-coinfected persons. Certainly one challenge has been studies of drug-drug interactions among these HCV protease inhibitors and ARVs. New information on the clinical pharmacology of these HCV agents was presented, which is a necessary, albeit a bit late, first step.
Boceprevir is a potent enzyme inhibitor, and is susceptible to induction and inhibition.
Information on the clinical pharmacology of BOC and several drug-drug interaction studies were presented in Abstract 118. The table below summarizes some of these interaction studies.
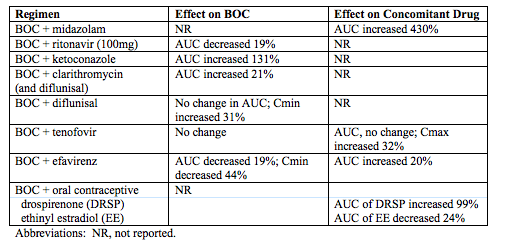
Here are my interpretations of these data.
· BOC is a potent inhibitor of CYP3A4. The evidence for this is the substantial increase in midazolam, which is a probe agent for this enzyme.
· BOC is not a substrate for CYP3A4/5. In preclinical metabolism studies, including experiments with RTV, BOC was found to be a substrate of CYP3A4/5 and RTV was a potent inhibitor of BOC metabolism (Antiviral Chem Chemother 2007;18:163-167). Therefore, BOC concentrations were expected to increase when it was given with RTV in these healthy volunteers, just as seen when RTV is given with HIV PIs that are CYP3A4/5 substrates. This increase didn't occur, however; the most likely explanation is that BOC isn't a 3A4/5 substrate.
· BOC may be a P-gp substrate. Ketoconazole and clarithromycin are considered CYP3A4 and P-gp inhibitors. While the concentrations of BOC were increased with co-administration, the magnitude of the increase was quite different. Because RTV (a much more potent CYP3A4 inhibitor) didn't increase BOC concentrations, the increases caused by ketoconazole and clarithromycin are not caused by CYP3A4 inhibition, and thus may be the result of P-gp inhibition or some other pathway.
· BOC may be a weak substrate for aldo keto reductase (AKR). AKRs are a family of enzymes that can play a role in the metabolism of drugs similar to the cytochrome P450 family of enzymes (CYPs). Preclinical metabolism studies indicated that BOC is substrate for certain AKR enzymes. Diflunisal is considered an inhibitor of AKRs, and coadministration with BOC would be expected to increase BOC concentrations. However, no change was seen in the AUC of BOC, although trough concentrations increased 31%. These data would suggest that either BOC is a weak substrate or diflunisal a weak inhibitor of AKR.
· EFV decreased the trough concentrations of BOC by 44%, presumably through CYP induction. EFV is a known inducer of CYP3A4 and 2B6. BOC, however, is not a substrate for CYP3A4 based on RTV interaction data. Interestingly, EFV and RTV had the same magnitude of effect on the AUC of BOC, both decreasing it by 19%. RTV is known to be an inducer of certain CYPs, such as CYP2C9, CYP2C19 and CYP1A2. EFV, in vitro, is actually an inhibitor of CYP2C9 and CYP2C19; EFV can also inhibit CYP1A2 but only at concentrations above those achieved clinically. The CYP enzyme EFV is inducing, which results in lower BOC concentrations, is not clear and needs to be determined because otherwise predicting enzyme induction drug interactions will be nearly impossible.
· Drospirenone (DRSP) in vitro is metabolized by CYP3A4 to a minor extent. As BOC is a potent inhibitor of CYP3A4 (see midazolam), inhibition may explain the increase in DRSP concentrations. A potential mechanism by which BOC would decrease ethinyl estradiol (EE) concentrations isn't obvious.
And, here are some take home points:
· There are differences between preclinical studies on BOC metabolism and the results of these drug-drug interactions in healthy volunteers. BOC appears not to be a substrate of CYP3A4/5. The authors of Abstract 118 (from Merck, Inc) also concluded that P-gp does not contribute to the elimination of BOC. Based on the ketoconazole data and the increase in both AUC and Cmax, I am not convinced that P-gp inhibition isn't playing a role in this interaction.
· BOC is going to be susceptible to certain drug interactions that can lower its concentrations as evidenced by the effect of EFV. I'll bet a dose increase of BOC will be required if given with EFV.
· BOC is going to have the potential to cause significant drug interactions for agents metabolized by CYP3A4 as evidenced by the substantial increase seen in midazolam concentrations.
· Collectively, these points lead me to my final comment on BOC - - there is a lot of clinical pharmacology work that needs to be done with this drug before it can be given with antiretrovirals and other concomitant medications that an HIV-infected person may take. In this regard, it was disappointing the authors of this abstract, who are the developers of this drug, came to CROI unprepared to discuss their plans for the drug-drug interaction studies required to move this agent forward in the HIV and HCV co-infected population.
Telaprevir (TVR) concentrations are not boosted by RTV, are decreased by EFV, and results with boosted PIs are mixed.
Abstract 629 evaluated the effect of low dose RTV on the PK of TVR in healthy volunteers. When the dose of TVR was decreased by 33% from 750 mg three times daily to 750 mg every 12 hours and given with RTV 100 mg every 12 hours, the trough concentration decreased by 32%. This corresponding decrease in TVR concentration with a decrease in TVR dose, despite the addition of RTV, indicates that RTV does not inhibit TVR metabolism and boost concentrations.
Drug-drug interaction studies with TVR and ARVs were reported in Abstract 119. The ARVs evaluated were: ATV/r, DRV/r, fAPV/r, LPV/r, EFV and TDF. In these studies the TVR dose was 750 mg every 8 hours, except for the study of TVR given with EFV and TDF, where because of the expected concentration-lowering effect of EFV, the dose of TVR was increased to 1125 mg every 8 hours.
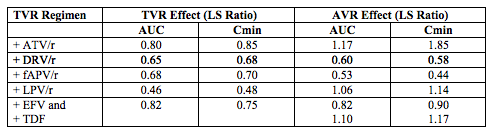
These data indicate all of the RTV-boosted PIs decreased TVR concentrations, ranging from a 20% decrease in AUC for ATV/r to a 54% decrease for LPV/r. The effect of TVR on the concentrations of the PIs, however, was mixed. TVR increased the AUC of ATV by 17%, while it decreased the AUC of APV by 47%. As expected, EFV reduced TVR concentrations.
These data indicate the most promising combination of TVR with a boosted PI that might not require dose adjustment is TVR and ATV/r. An increase in the TVR dose is necessary if given with EFV and an increase to 1125 mg every 8 hours appeared to overcome some of the lowering effect of EFV, but AUC and Cmin values were not completely normalized. Based on these results, a Phase 2 study was initiated and interim results were presented in Abstract 146LB.
Interim results of a TVR-containing HCV regimen in HIV and HCV co-infected persons receiving an ATV/r or EFV based ARV regimen are encouraging.
In this Phase 2 study of HIV and HCV co-infected persons, TVR was given without ART, or with a regimen of ATV/r or EFV in combination with TDF/FTC. In addition to TVR the HCV regimen for the first 12 weeks included pegylated interferon and ribavirin. The table below gives the percent of participants who had undetectable HCV RNA at weeks 4 and 12.

These results are indeed encouraging showing an HCV regimen containing TVR can be given safely with an ARV regimen, achieve an HCV response and not compromise HIV suppression in the participants who were receiving ART. It is noticeable the HCV response in the cohort who received TVR with ATV/r is lower than no ART or EFV. When this question was asked of the presenter, the response was this is likely an effect of a small sample size, and this may be the case. However, no one can yet be confident this is not an effect of the drug interaction between ATV/r and TVR where TVR concentrations are decreased. That drug interaction study has a small sample size as well; therefore, the lowering effect could be larger. Additionally, the PK results of drug interactions in patients may not look like those in healthy volunteers. So, I strongly suggest everyone keep an open mind as to whether the TVR doses used in this Phase 2 study are the "optimal" doses necessary to manage the drug-drug interactions between TVR and ATV/r, and TVR and EFV.
And elsewhere in HIV therapeutics: once daily raltegravir is inferior to twice daily, and a PK/PD relationship does exist.
Depending on your perspective the results of the raltegravir once daily study (abstract 150LB) may or may not be an advance in therapeutics. This non inferiority trial of once vs. twice daily RAL in 770 treatment-naive persons demonstrated once daily therapy was inferior to twice daily, with overall proportions of subjects with HIV-RNA <50cpm at week 48 of 83.2 vs. 88.9, respectively. Nine subjects receiving once daily compared with 2 receiving twice daily developed integrase-resistant virus. Pharmacokinetic data reported at the conference showed the AUC of once vs. twice-daily RAL were similar, as would be expected from giving the same total daily dose. However, the trough concentrations with once daily were substantially lower: the geometric mean of all troughs was 83nM for once daily but was 380nM for twice. Furthermore, a higher trough was associated with a greater probability of successful treatment outcome.
These data should end the nonsensical statements of "there is no PK/PD relationship for RAL". Even without these data this statement was wrong. There was always such a relationship; it was simply that concentrations with the twice-daily dose were sufficiently high on the concentration-response curve the quantitative nature of it was obscured. The trough concentrations achieved with once daily dosing on the other hand fell off of the flat, plateau phase and revealed that low troughs were associated with virology failure. In this regard, RAL is just like the integrase inhibitors elvitegravir and dolutegravir (S/GSK1349572) where clear relationships have been described between trough concentrations and virologic response. This trial doesn't support use of once daily RAL for initiation of therapy in the treatment-naive person. But it does close some gaps in our knowledge of the clinical pharmacology of this drug, and if we learn the right lessons should prevent other therapeutic misadventures.
II. Drug Interactions
Rifabutin pharmacokinetics when given with LPV/r support the need for a higher dose.
The most important drug-drug interaction study at this meting was a study from South Africa that examined rifabutin dosing regimens when given with LPV/r in HIV-infected persons (abstract 650). The current recommendation is to use a rifabutin regimen of 150mg three times weekly if given with a boosted PI. However, concern has been increasing whether this is an appropriate dose because of reports of emergence of rifamycin resistance and that this dose adjustment approach is based on pharmacokinetic studies conducted in healthy volunteers and has not been rigorously confirmed in HIV-infected persons. Pym and colleagues determined rifabutin pharmacokinetics following the standard dose of 300mg once daily, and after doses of 150mg three times weekly (TIW) and 150mg once daily given with LPV/r twice daily (Abstract 650). The following table gives the rifabutin parameters for these three doses.
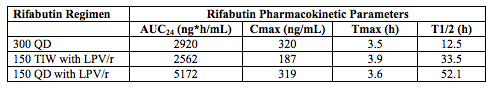
Rifabutin Cmax is generally considered the pharmacokinetic characteristic related to response. These data indicate when rifabutin is given with LPV/r the once daily dose achieves Cmax values comparable to that of the usual, 300mg once daily dose, whereas the presently recommended 150mg TIW dose achieves Cmax values almost half. These data provide a pharmacokinetic basis to support increasing the rifabutin dose when given with LPV/r to 150mg per day. Clinical confirmation of the safety and efficacy of this dose is needed, and soon.
Enzyme inducing anti-epileptics may increase the risk of virologic failure.
Abstract 646 was an important reminder that drugs less commonly used in developed countries, may still be frequently used in resource limited setting and may be associated with significant interactions with ARVs. Patients in the US Military HIV Natural History Study taking a PI or NNRTI-based ARV regimen for more than 6 months with anti-epileptics for at least 28 days were evaluated for their risk of virologic failure. Anti-epileptics were categorized as enzyme inducing (phenytoin, carbamazepine and phenobarbital) and non-enzyme inducing (e.g. lamotrigine, levetiracetam and gabapentin). Recipients of the enzyme inducing anti-epileptics has a significantly greater risk of virologic failure than those not receiving enzyme inducing agents: 10/17 (59%) versus 20/75 (27%); odds ratio, 3.9. These data illustrate an increased risk of virologic failure associated with enzyme inducing anti-epileptics. If at all possible, concomitant administration of these drugs should be avoided. If not possible, this would be a scenario for therapeutic drug monitoring, if available.
III. Compartmental Penetration
A new prodrug of TFV is more potent and achieves higher intracellular concentrations than TDF.
Tenofovir disoproxil fumarate (TDF) is a prodrug of tenofovir (TFV). That is, TDF is taken orally and converted in the blood stream to TFV that is then converted intracellularly to TFV-diphosphate, which is the pharmacologically-active drug. Abstract 152LB described a new prodrug of TFV-diphosphate, GS-7340, which is more potent and achieves higher intracellular concentrations than does TDF. The in vitro potency as measured by the IC50 for TFV is 1.2 μM, is 0.015 μM for TDF, and is 0.003 μM for GS-7340. TDF (300 mg once daily) and GS-7340 (50 or 150 mg once daily) were given to treatment-naïve, HIV-infected volunteers for 14 days. The time-weighted change in viral load was -0.54 for TDF, -0.95 for 50 mg GS-7340, and -1.07 for 150 mg of GS-7340. Compared with TDF, the plasma concentrations of TFV were approximately 90% lower and the intracellular PBMC concentrations were variable but some 30-fold higher with 150 mg of GS-7340. There was no difference in short-term safety. GS-7340 represents a very interesting strategy: a prodrug that achieves lower plasma concentrations, so hopefully fewer adverse effects, but higher intracellular concentrations and hopefully improved long-term suppression of HIV. Because GS-7340 requires a lower dose of TFV to achieve a greater suppression of HIV, the cost of goods should be lower, so perhaps (hopefully) it will be less expensive? An intriguing approach - - stay tuned.
Concentrations of darunavir and etravirine in the CSF appear sufficient to inhibit HIV.
The CHARTER Group continues its effort to provide information on the penetration of ARVs into the CSF. In Abstract 643, they report on CSF concentrations of DRV and ETR. The median CSF-to-plasma ratio for DRV was 0.014 and for ETR was 0.042. The CSF concentrations for both drugs were greater than the IC50 of wild-type HIV in all of the participants. These data suggest that DRV and ETR penetrate the CSF in levels that may contribute to suppression of HIV in the CSF.
IV. Adverse Drug Reactions - Complications of ARV Use
No association found with abacavir (ABC) use and MI in FDA meta-analysis.
The association between abacavir use and myocardial infarction (MI) is back in the news with a meta-analysis presented by the FDA (Abstract 808). They analyzed data from 26 randomized controlled trials that included 9832 subjects with a mean follow-up of 1.43 person years in the ABC group and 1.49 person years in the non-ABC group. There was no significant difference between these groups and the risk of MI: risk difference, 0.03% (95% CI, -0.24 to 0.30%). So, where do we stand. This analysis sure seems to raise uncertainty about ABC use and an increased risk of MI. But, how does the strength of this finding of no association with 1.48 years of person follow-up and 46 MI events, compare with the strength of the finding of a positive association with MI in the D:A:D observational cohort whose most recent report had 5.8 person years of follow-up and 580 MI events (J Infect Dis 2010; 201: 318-30)? I agree with the final conclusion in this abstract, that the only way to understand a cardiovascular risk of ABC use is to conduct a randomized, prospective study with CV events as a primary endpoint. Who is going to do that study?
CROI 2011 Pcol-NATAP.doc
|
| |
|
|
|
|
|