| |
Approval of INCIVEK (telaprevir), a direct acting antiviral drug (DAA) to treat hepatitis C (HCV): stopping rules. Study results (naives, treatment-experienced) (FDA Announcement)
|
| |
| |
(FDA) On May 23, 2011, FDA approved INCIVEK (telaprevir), a hepatitis C virus (HCV) protease inhibitor. INCIVEK is the second direct acting antiviral drug against the hepatitis C virus to be approved.
INCIVEK (telaprevir), in combination with peginterferon alfa and ribavirin, is indicated for the treatment of genotype 1 chronic hepatitis C in adult patients with compensated liver disease, including cirrhosis, who are treatment-naïve (patients who have not received interferon-based drug therapy for their infection) or who have previously been treated with interferon-based treatment and not responded adequately, including prior null responders, partial responders, and relapsers.
The current standard of care for patients with hepatitis C infection is peginterferon alfa and ribavirin taken for 48 weeks. Less than 50 percent of patients respond to this therapy.
The following points should be considered when initiating treatment with INCIVEK:
· INCIVEK must not be administered as monotherapy and must only be prescribed with both peginterferon alfa and ribavirin.
· A high proportion of previous null responders (particularly those with cirrhosis) did not achieve a Sustained Virologic Response (SVR) and had telaprevir resistance-associated substitutions emerge on treatment with INCIVEK combination treatment.
· INCIVEK efficacy has not been established for patients who have previously failed therapy with a treatment regimen that includes INCIVEK or other HCV NS3/4A protease inhibitors
DOSAGE AND ADMINISTRATION
INCIVEK/Peginterferon Alfa/Ribavirin Combination Treatment
The recommended dose of INCIVEK tablets is 750 mg (two 375-mg tablets) taken orally 3 times a day (7-9 hours apart) with food (not low fat).
For specific dosage instructions for peginterferon alfa and ribavirin, refer to their respective prescribing information.
Duration of Treatment
The recommended duration of treatment with INCIVEK is 12 weeks in combination with peginterferon alfa and ribavirin. HCV-RNA levels should be monitored at weeks 4 and 12 to determine combination treatment duration and assess for treatment futility (Tables 1 and 2).
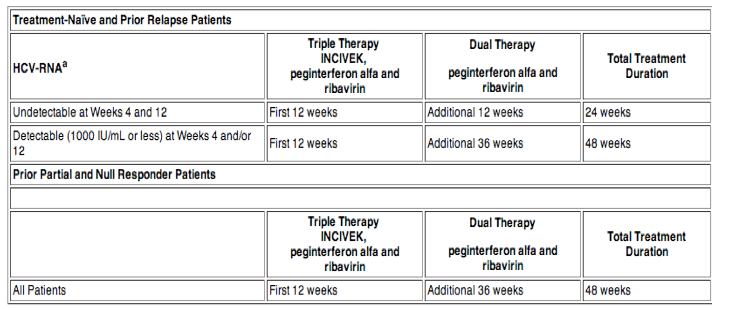
aIn clinical trials, HCV-RNA in plasma was measured using a COBAS® TaqMan® assay with a lower limit of quantification of 25 IU/mL and a limit of detection of 10 IU/mL.
For the purpose of assessing response-guided therapy eligibility at weeks 4 and 12 (see Table 1), an "undetectable" HCV-RNA result is required; a confirmed "detectable but below limit of quantification" HCV-RNA result should not be considered equivalent to an "undetectable" HCV-RNA result.
Treatment-naïve patients with cirrhosis who have undetectable HCV-RNA at weeks 4 and 12 of INCIVEK combination treatment may benefit from an additional 36 weeks of peginterferon alfa and ribavirin (48 weeks total)
Dose Reduction
To prevent treatment failure, the dose of INCIVEK must not be reduced or interrupted. Refer to the respective prescribing information for dose modification of peginterferon alfa and ribavirin.
Discontinuation of Dosing
Patients with inadequate viral response are unlikely to achieve SVR, and may develop treatment-emergent resistance substitutions. Discontinuation of therapy is recommended in all patients with (1) HCV-RNA levels of greater than or equal to 1000 IU/mL at Treatment Week 4 or 12; or (2) confirmed detectable HCV-RNA levels at Treatment Week 24 (see Table 2).

CLINICAL TRIAL RESULTS
The approval of INCIVEK is based on safety and efficacy data in approximately 2250 adult subjects who were previously untreated (ADVANCE and ILLUMINATE) or who had failed previous peginterferon alfa and ribavirin therapy (REALIZE) in clinical studies
Treatment-Naïve Adults
Study 108 (ADVANCE)
Study 108 was a randomized, double-blind, parallel-group, placebo-controlled, trial conducted in treatment-naïve subjects (had received no prior therapy for HCV, including interferon or pegylated interferon monotherapy). INCIVEK was given for the first 8 weeks of treatment (T8/PR regimen) or the first 12 weeks of treatment (T12/PR regimen) in combination with Peg-IFN-alfa-2a/RBV for either 24 or 48 weeks. Subjects who had undetectable HCV-RNA at weeks 4 and 12 (extended Rapid Virologic Response [eRVR]) received 24 weeks of Peg-IFN-alfa-2a/RBV treatment, and subjects who did not have undetectable HCV-RNA at weeks 4 and 12 (no eRVR) received 48 weeks of Peg-IFN-alfa-2a/RBV treatment. The control regimen (Pbo/PR48) had a fixed treatment duration, with telaprevir matching placebo for the first 12 weeks and Peg-IFN-alfa-2a/RBV for 48 weeks.
The 1088 enrolled subjects had a median age of 49 years (range: 18 to 69); 59% of the subjects were male; 23% had a body mass index greater than or equal to 30 kg/m2; 9% were Black; 11% were Hispanic or Latino; 77% had baseline HCV-RNA levels greater than 800,000 IU/mL; 15% had bridging fibrosis; 6% had cirrhosis; 59% had HCV genotype 1a; and 40% had HCV genotype 1b.
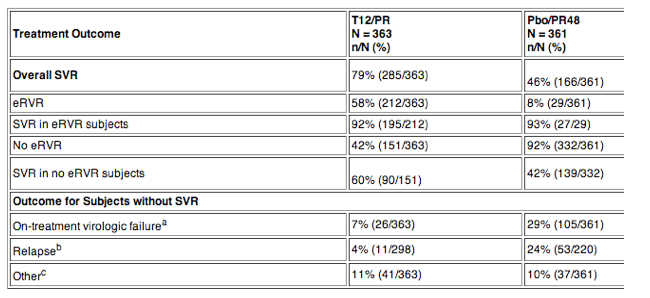
a On-treatment failure includes subjects who met a protocol-defined virologic stopping rule or who had detectable HCV-RNA at the time of their last dose of INCIVEK and subjects who had viral breakthrough on peginterferon alfa/ribavirin.
b Relapse rates are calculated with a denominator of subjects with undetectable HCV-RNA at the end of treatment.
c Other includes subjects with detectable HCV-RNA at the time of their last study drug but who did not have viral breakthrough, and subjects with a missing SVR assessment.
In the T8/PR group, the overall SVR rate was 72%. The eRVR rate was 57% and the SVR rate for eRVR subjects was 87%. The SVR rate for no eRVR subjects was 52%. More subjects in the T8/PR group experienced virologic breakthrough after Week 12 while receiving peginterferon alfa and ribavirin alone, 16% compared to 10% in T12/PR group.
SVR rates were higher (absolute difference of at least 22%) for the T12/PR group than for the Pbo/PR48 group across subgroups by sex, age, race, ethnicity, body mass index, HCV genotype subtype, baseline HCV-RNA (less than 800,000, greater than or equal to 800,000 IU/mL), and extent of liver fibrosis. However, there were small numbers of subjects enrolled in some key subgroups. In the T12/PR group:
· Twenty-one subjects had cirrhosis at baseline and the overall SVR in these subjects was 62% (13/21). Among subjects with cirrhosis, 43% (9/21) achieved an eRVR and of those 78% (7/9) achieved SVR.
· Twenty-six subjects were Black/African Americans. The overall SVR among Black/African American subjects was 62% (16/26). Among these subjects, 31% (8/26) achieved an eRVR and of those 89% (8/9) achieved SVR.
Study 111 (ILLUMINATE)
Study 111 was a randomized, open label trial conducted in treatment naïve subjects. The study was designed to compare SVR rates in subjects achieving eRVR who were treated with INCIVEK for 12 weeks in combination with Peg-IFN-alfa-2a/RBV for either 24 weeks (T12/PR24 regimen) or 48 weeks (T12/PR48 regimen).
The 540 enrolled subjects had a median age of 51 years (range: 19 to 70); 60% were male; 32% had a body mass index greater than or equal to 30 kg/m2; 14% were Black; 10% were Hispanic or Latino; 82% had baseline HCV-RNA levels greater than 800,000 IU/mL; 16% had bridging fibrosis; 11% had cirrhosis; 72% had HCV genotype 1a; and 27% had HCV genotype 1b.
The SVR rate for all subjects enrolled in the trial was 74%. A total of 352 (65%) subjects achieved eRVR and of those 322 (60%) were randomized to 24 weeks (T12/PR24, n=162) or 48 weeks (T12/PR48, n=160) of treatment. The SVR rates were similar at 92% (T12/PR24) and 90% (T12/PR48), respectively. Again, small numbers of subjects were enrolled in some key subgroups:
· Sixty-one (11%) of subjects had cirrhosis at baseline. Among subjects with cirrhosis, 30 (49%) achieved an eRVR: 18 were randomized to T12/PR24 and 12 to T12/PR48. The SVR rates were 67% (12/18) for the T12/PR24 group and 92% (11/12) for the T12/PR48 group.
· Blacks/African Americans comprised 14% (73/540) of study subjects. Thirty-four (47%) Black/African American subjects achieved an eRVR and were randomized to T12/PR24 or T12/PR48. The respective SVR rates were 88% (15/17) and 94% (16/17), compared to 93% (246/265) for Caucasians.
Previously Treated Adults
Study C216 (REALIZE)
Study C216 was a randomized, double-blind, placebo-controlled, trial conducted in subjects who did not achieve SVR with prior treatment with Peg-IFN-alfa-2a/RBV or Peg IFN alfa-2b/RBV. The study enrolled prior relapsers (subjects with HCV-RNA undetectable at end of treatment with a pegylated interferon-based regimen, but HCV RNA detectable within 24 weeks of treatment follow-up) and prior non responders (subjects who did not have undetectable HCV-RNA levels during or at the end of a prior course of at least 12 weeks of treatment). The nonresponder population included 2 subgroups: prior partial responders (greater than or equal to 2 log10 reduction in HCV-RNA at week 12, but not achieving HCV RNA undetectable at end of treatment with peginterferon alfa and ribavirin) and prior null responders (less than 2 log10 reduction in HCV-RNA at week 12 of prior treatment with peginterferon alfa and ribavirin).
Subjects were randomized in a 2:2:1 ratio to one of two INCIVEK combination treatment groups (with and without a Peg-IFN-alfa-2a/RBV lead in) or a control group. The T12/PR48 group received INCIVEK and Peg-IFN-alfa-2a/RBV for 12 weeks (without a lead-in), followed by placebo and Peg-IFN-alfa-2a/RBV for 4 weeks, followed by Peg-IFN-alfa-2a/RBV for 32 weeks. The T12(DS)/PR48 group had a lead-in (delayed start of INCIVEK) with placebo and Peg-IFN-alfa-2a/RBV for 4 weeks, followed by INCIVEK and Peg IFN alfa-2a/RBV for 12 weeks, followed by Peg-IFN-alfa-2a/RBV for 32 weeks. The Pbo/PR48 group received placebo and Peg-IFN-alfa-2a/RBV for 16 weeks, followed by Peg-IFN-alfa-2a/RBV for 32 weeks.
The 662 enrolled subjects had a median age of 51 years (range: 21 to 70); 70% of the subjects were male; 26% had a body mass index greater than or equal to 30 kg/m2; 5% were Black; 11% were Hispanic or Latino; 89% had baseline HCV-RNA levels greater than 800,000 IU/mL; 22% had bridging fibrosis; 26% had cirrhosis; 54% had HCV genotype 1a, and 46% had HCV genotype 1b. Null and partial responders had higher baseline HCV-RNA levels and more advanced liver disease (cirrhosis) than relapsers; other characteristics were similar across these populations.
The lead-in and immediate start regimens produced comparable SVR and no SVR rates, so data from these two groups were pooled (Table 11).

a Lead-in and immediate start T12/PR regimens pooled
b On-treatment virologic failure includes subjects who met a protocol-defined virologic stopping rule or who had detectable HCV-RNA at the time of their last dose of INCIVEK and subjects who had viral breakthrough on peginterferon alfa/ribavirin.
c Relapse rates are calculated with a denominator of subjects with undetectable HCV-RNA at the end of treatment.
Among prior relapsers, 76% (218/286) achieved an eRVR and of those 95% (208/218) achieved an SVR. In an earlier, dose-finding clinical trial, 78% (52/67) of prior relapsers achieved an eRVR and were treated with 24 weeks of peginterferon alfa and ribavirin (T12/PR24); of those 94% (49/52) achieved an SVR.
For all populations in the study (prior relapsers, prior partial responders, and prior null responders), SVR rates were higher for the T12/PR group than for the Pbo/PR48 group across subgroups by sex, age, ethnicity, body mass index, HCV genotype subtype, baseline HCV-RNA level, and extent of liver fibrosis.
Twenty-three percent of INCIVEK-treated subjects had cirrhosis at baseline. SVR rates among cirrhotic subjects who received INCIVEK combination treatment compared to Pbo/PR48 were: 87% (48/55) compared to 13% (2/15) for prior relapsers, 34% (11/32) compared to 20% (1/5) for prior partial responders, and 14% (7/50) compared to 10% (1/10) for prior null responders.
Four percent (19/530) of treatment experienced subjects who received INCIVEK combination treatment were Black/African Americans; the SVR rate for these subjects was 63% (12/19) compared to 65% (328/503) for Caucasians.
CONTRAINDICATIONS
INCIVEK combination treatment is contraindicated in:
· Pregnant women and men whose female partners are pregnant because of the risks for birth defects and fetal death associated with ribavirin
· INCIVEK is contraindicated when combined with drugs that are highly dependent on CYP3A for clearance and for which elevated plasma concentrations are associated with serious and/or life-threatening events (narrow therapeutic index). INCIVEK is contraindicated when combined with drugs that strongly induce CYP3A and thus may lead to lower exposure and loss of efficacy of INCIVEK. The contraindicated medications include the following:
Alfuzosin
Rifampin
Dihydroergotamine, ergonovine, ergotamine, methylergonovine
Cisapride
St John's wort (Hypericum perforatum)
Atorvastatin, lovastatin, simvastatin
Pimozide
Sildenafil (Revatio®) or tadalafil (Adcirca®) [for treatment of pulmonary arterial hypertension]
· WARNINGS AND PRECAUTIONS
The Warnings and Precautions for INCIVEK include drug interactions and the following:
Pregnancy (Use with Ribavirin and Peginterferon Alfa)
· Ribavirin may cause birth defects and fetal death; avoid pregnancy in female patients and female partners of male patients. Patients must have a negative pregnancy test prior to therapy and have monthly pregnancy test and for six months after treatment ends. Hormonal contraceptives may not be reliable during INCIVEK dosing and for up to two weeks following cessation of INCIVEK During this time, female patients of childbearing potential should use 2 non-hormonal methods of effective birth control. Examples of non-hormonal methods of contraception include a male condom with spermicidal jelly OR female condom with spermicidal jelly (a combination of a male condom and a female condom is not suitable), a diaphragm with spermicidal jelly, a cervical cap with spermicidal jelly, or an intrauterine device (IUD).
Serious Skin Reactions
· Serious skin reactions, including Drug Rash with Eosinophilia and Systemic Symptoms (DRESS) and Stevens-Johnson Syndrome (SJS) were reported in less than 1% of subjects who received INCIVEK combination treatment compared to none who received peginterferon alfa and ribavirin alone. These serious skin reactions required hospitalization, and all patients recovered. The presenting signs of DRESS may include rash, fever, facial edema, and evidence of internal organ involvement (e.g., hepatitis, nephritis). Eosinophilia may or may not be present. The presenting signs of SJS may include fever, target lesions, and mucosal erosions or ulcerations (e.g., conjunctivae, lips).
If a serious skin reaction occurs, all components of INCIVEK combination treatment must be discontinued immediately and the patient should be promptly referred for urgent medical care
Rash
Rash developed in 56% of subjects who received INCIVEK combination treatment [see Adverse Reactions (6.1)]. Severe rash (e.g., a generalized rash or rash with vesicles or bullae or ulcerations other than SJS) was reported in 4% of subjects who received INCIVEK combination treatment compared to less than 1% who received peginterferon alfa and ribavirin alone. The severe rash may have a prominent eczematous component.
Patients with mild to moderate rashes should be followed for progression of rash or development of systemic symptoms. If rash progresses and becomes severe or if systemic symptoms develop, INCIVEK should be discontinued. Peginterferon alfa and ribavirin may be continued. If improvement is not observed within 7 days of INCIVEK discontinuation, sequential or simultaneous interruption or discontinuation of ribavirin and/or peginterferon alfa should be considered. If medically indicated, earlier interruption or discontinuation of ribavirin and peginterferon alfa should be considered. Patients should be monitored until the rash has resolved. INCIVEK must not be reduced or restarted if discontinued due to rash. Treatment of rash with oral antihistamines and/or topical corticosteroids may provide symptomatic relief but effectiveness of these measures has not been established. Treatment of rash with systemic corticosteroids is not recommended [see Drug Interactions (7)].
Anemia
· Anemia has been reported with peginterferon alfa and ribavirin therapy. The addition of INCIVEK to peginterferon alfa and ribavirin is associated with an additional decrease in hemoglobin concentrations. Hemoglobin values less than or equal to 10 g/dL were observed in 36% of subjects who received INCIVEK combination treatment compared to 17% of subjects who received peginterferon alfa and ribavirin. Hemoglobin values less than 8.5 g/dL were observed in 14% of subjects who received INCIVEK combination treatment compared to 5% of subjects receiving peginterferon alfa and ribavirin.
· In subjects receiving INCIVEK combination treatment, 4% discontinued INCIVEK, 1% discontinued INCIVEK combination treatment, and 32% underwent a ribavirin dose modification (reduction, interruption or discontinuation) due to anemia. In subjects treated with peginterferon alfa and ribavirin alone, there were two discontinuations and 12% underwent ribavirin dose modification due to anemia.
· Hemoglobin should be monitored prior to and at least every 4 weeks during INCIVEK combination treatment. For the management of anemia, ribavirin dose reductions should be used (refer to the prescribing information for ribavirin for its dose reduction guidelines). If ribavirin dose reductions are inadequate, discontinuation of INCIVEK should be considered. If ribavirin is permanently discontinued for the management of anemia, INCIVEK must also be permanently discontinued. Ribavirin may be restarted per the dosing modification guidelines for ribavirin. The dose of INCIVEK must not be reduced and INCIVEK must not be restarted if discontinued.
Laboratory Tests
· HCV-RNA levels should be monitored at weeks 4 and 12 and as clinically indicated. Use of a sensitive real-time RT-PCR assay for monitoring HCV-RNA levels during treatment is recommended. The assay should have a lower limit of HCV-RNA quantification equal to or less than 25 IU/mL and a limit of HCV-RNA detection of approximately 10-15 IU/mL. For the purpose of assessing response-guided therapy eligibility, an "undetectable" HCV-RNA result is required; a confirmed "detectable but below limit of quantification" HCV-RNA result should not be considered equivalent to an "undetectable" HCV-RNA result.
Hematology evaluations (including white cell differential count) are recommended at weeks 2, 4, 8 and 12 or as clinically appropriate thereafter.
Chemistry evaluations (electrolytes, serum creatinine, uric acid, hepatic enzymes, bilirubin, and TSH) are recommended as frequently as the hematology evaluations or as clinically indicated
General
· INCIVEK must not be administered as monotherapy and must only be prescribed with both peginterferon alfa and ribavirin. Therefore, the prescribing information for peginterferon alfa and ribavirin must be consulted before starting treatment with INCIVEK.
There are no clinical data on re-treating patients who have failed an HCV NS3/4A protease inhibitor-based treatment, nor are there data on repeated courses of INCIVEK [see Microbiology (12.4)].
Hepatic Impairment
· INCIVEK is not recommended for patients with moderate or severe hepatic impairment (Child-Pugh B or C, score greater than or equal to 7) or patients with decompensated liver disease.
· ADVERSE DRUG REACTIONS
The most commonly reported adverse reactions in adult subjects were rash, fatigue, pruritus, nausea, anemia, diarrhea, vomiting, hemorrhoids, anorectal discomfort, dysgeusia and anal pruritis.
Serious adverse drug reactions occurred in 3% of subjects who received INCIVEK combination treatment compared to none of the subjects treated with peginterferon alfa and ribavirin. The most frequent serious adverse events in subjects treated with INCIVEK combination treatment were skin disorders (rash and/or pruritus) and anemia. Fourteen percent of subjects discontinued INCIVEK due to adverse drug reactions. Rash, anemia, fatigue, pruritus, nausea, and vomiting were the most frequent adverse drug reactions leading to discontinuation of INCIVEK
·
USE IN SPECIAL POPULATIONS
- Hepatic Impairment: Safety and efficacy have not been established in patients with Child-Pugh score greater than or equal to 7 (class B and C)
- Co-infection: Safety and efficacy have not been established in HCV/HIV and HCV/HBV co-infected patients.
- Pediatrics: Safety and efficacy have not been established in pediatric patients.
- Solid Organ Transplant: Safety and efficacy have not been established in patients undergoing solid organ transplants
- Ribavirin Pregnancy Registry available
Telaprevir is a product of Vertex Pharmaceuticals Incorporated.
The complete product label will be available soon on the FDA web site at Drugs@FDA.
Richard Klein
Office of Special Health Issues
Food and Drug Administration
Kimberly Struble
Division of Antiviral Drug Products
Food and Drug Administration
|
|
| |
| |
|
|
|