| |
Hepatitis: Brain endothelial cells support HCV entry and replication
|
| |
| |
Download the PDF here
Nature Reviews Gastroenterology and Hepatology, advance online publication, Published online 17 January 2012
Isobel Franks
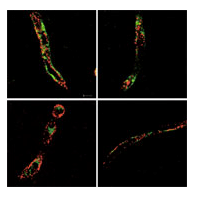
HCV receptor expression in human brain tissue. Courtesy of J. McKeating.
Nicola Fletcher, Jane McKeating and colleagues have demonstrated that human brain endothelial cells express all the main receptors required for HCV entry. "This is the first report that cells of the central nervous system (CNS) support HCV entry and replication in vitro," says McKeating.
Chronic HCV infection causes progressive liver disease and can also be associated with various CNS abnormalities. To date, however, few studies have investigated the mechanisms by which these abnormalities arise, that is, whether they are a result of impaired hepatic function or virus replication in the CNS. Thus, the researchers quantified HCV RNA levels in the brain and liver of 10 individuals infected with HCV. They also investigated the expression of HCV entry receptors in the brain, and conducted in vitro studies to ascertain whether two brain-derived endothelial cell lines could support HCV infection.
HCV RNA was detected in multiple brain samples from four of the patients (although at much lower levels than in the liver samples), and microvascular endothelia expressed all of the essential HCV entry receptors. The two cell lines supported HCV entry and replication; infection was inhibited by receptor-specific antibodies, interferon and antiviral agents. Furthermore, infection caused endothelial cell apoptosis, potentially providing a mechanism for the mild neurocognitive impairment observed in patients with chronic hepatitis C.
"We have now demonstrated that HCV can infect brain endothelial cells, providing the first model to study HCV neuropathology," concludes McKeating. The researchers also suspect that brain endothelial cells might provide a viral reservoir during antiviral treatment.
-------
Hepatitis C Virus Infects the Endothelial Cells of the Blood-Brain Barrier - pdf attached
Gastroenterology Nov 2011
NICOLA F. FLETCHER,* GARRICK K. WILSON,* JACINTA MURRAY,ŕ KE HU,* ANDREW LEWIS,¤ GARY M. REYNOLDS,* ZANIA STAMATAKI,* LUKE W. MEREDITH,* IAN A. ROWE,* GUANGXIANG LUO, MIGUEL A. LOPEZ-RAMIREZ,¦ THOMAS F. BAUMERT,# BABETTE WEKSLER,** PIERRE-OLIVIER COURAUD,ŕŕ KWANG SIK KIM,¤¤ IGNACIO A. ROMERO,¦ CATHERINE JOPLING,¤ SUSAN MORGELLO,ŕ PETER BALFE,* and JANE A. McKEATING*
*Hepatitis C Research Group, Institute for Biomedical Research, University of Birmingham, Birmingham, England; ŕDepartment of Pathology, Mount Sinai School of Medicine, New York, New York; ¤School of Pharmacy, University of Nottingham, Nottingham, England; Department of Microbiology, Immunology and Molecular Genetics, University of Kentucky, Lexington, Kentucky; ¦Department of Life Sciences, The Open University, Milton Keynes, England; #UniversitŽ de Strasbourg and P™le HŽpato-digestif, H™pitaux Universitaires de Strasbourg, Strasbourg, France; **Weill Cornell Medical College, New York, New York; ŕŕInstitut Cochin, CNRS UMR 8104, INSERM UnitŽ 567, UniversitŽ Paris Descartes, Paris, France; and ¤¤Division of Infectious Diseases, The Johns Hopkins School of Medicine, Baltimore,
Maryland
Received 18 June 2011; accepted 15 November 2011. published online 02 December 2011.
Uncorrected Proof
"We measured levels of HCV RNA and expression of the viral entry receptor in brain tissue samples from 10 infected individuals (and 3 uninfected individuals, as controls) and human brain microvascular endothelial cells......Using quantitative polymerase chain reaction, we detected HCV RNA in brain tissue of infected individuals at significantly lower levels than in liver samples. Brain microvascular endothelia and brain endothelial cells expressed all of the recognized HCV entry receptors.......Virus infection of the CNS might lead to HCV-associated neuropathologies.......Quantification of HCV RNA from matched samples of white and grey matter, cerebellum, medulla, liver, and plasma revealed that, in clinical samples with detectable brain HCV, the viral load was 1000- to 10,000-fold lower in brain compared with liver from the same subject. HCV RNA was detected in at least one brain region from 4 of 10 HCV-infected subjects, independent of HIV coinfection status.......It is worth noting that 6 HCV-infected subjects had no detectable viral RNA in the brain, despite having comparable levels of viral RNA in the liver and plasma (Supplementary Materials and Methods) to 4 subjects with no detectable HCV RNA in the brain........In conclusion, we show that brain microvascular endothelium expresses all the major viral receptors required for HCV infection. Two brain endothelial cell lines support HCV entry and replication, in which infection is inhibited by HCV receptor-specific antibodies, interferon, and specific antiviral agents. The observation that HCV-infected hCMEC/D3 cells release low levels of infectious virus and show evidence of apoptosis supports a model in which the BBB may provide an extrahepatic target for infection, and HCV may directly induce neuropathology in vivo........these data show that brain endothelial cells release low levels of HCV that are infectious for hepatoma cells......HCV infection affects hCMEC/D3 paracellular permeability....showing a direct effect of infection on brain endothelial cell apoptosis......HCV infection leads to progressive liver disease, which has been associated with extrahepatic syndromes, including CNS abnormalities.3 "
Background & Aims
Hepatitis C virus (HCV) infection leads to progressive liver disease and is associated with a variety of extrahepatic syndromes, including central nervous system (CNS) abnormalities. However, it is unclear whether such cognitive abnormalities are a function of systemic disease, impaired hepatic function, or virus infection of the CNS.
Methods
We measured levels of HCV RNA and expression of the viral entry receptor in brain tissue samples from 10 infected individuals (and 3 uninfected individuals, as controls) and human brain microvascular endothelial cells by using quantitative polymerase chain reaction and immunochemical and confocal imaging analyses. HCV pseudoparticles and cell culture-derived HCV were used to study the ability of endothelial cells to support viral entry and replication.
Results
Using quantitative polymerase chain reaction, we detected HCV RNA in brain tissue of infected individuals at significantly lower levels than in liver samples. Brain microvascular endothelia and brain endothelial cells expressed all of the recognized HCV entry receptors. Two independently derived brain endothelial cell lines, hCMEC/D3 and HBMEC, supported HCV entry and replication. These processes were inhibited by antibodies against the entry factors CD81, scavenger receptor BI, and claudin-1; by interferon; and by reagents that inhibit NS3 protease and NS5B polymerase. HCV infection promotes endothelial permeability and cellular apoptosis.
Conclusions
Human brain endothelial cells express functional receptors that support HCV entry and replication. Virus infection of the CNS might lead to HCV-associated neuropathologies.
Hepatitis C virus (HCV) is an enveloped positive-strand RNA virus classified in the Hepacivirus genus of the Flaviviridae family. Worldwide, approximately 170 million individuals are infected with HCV that leads to a progressive liver disease. Infection is associated with a variety of extrahepatic syndromes, including cryoglobulinemia, glomerulonephritis, and central nervous system (CNS) abnormalities.1
Although HCV is primarily a hepatotropic virus, genomic viral RNA has been detected in peripheral blood mononuclear cells, cerebrospinal fluid, and the brain of chronically infected patients with neuropathologic abnormalities (reviewed in Morgello2 and Weissenborn et al3). At present, there is no small animal model to study HCV pathobiology and studies on tropism are limited to humans. Analysis of HCV sequences derived from peripheral blood mononuclear cells, brain, and liver show tissue-specific differences, suggesting independent evolution at different anatomic sites.4, 5, 6
Virus tropism is likely to be defined at multiple stages of the viral life cycle, including entry, replication, and assembly. The availability of retroviral pseudoparticles bearing HCV glycoproteins (HCVpp) and the recently reported JFH-1 strain of HCV that replicates and assembles infectious particles in cell culture (HCVcc) have enabled considerable advances in our understanding of the receptors involved in HCV internalization.7, 8 Recent evidence shows a number of host cell molecules to be important for HCV entry: low-density lipoprotein receptor (LDL-R), tetraspanin CD81, scavenger receptor class B member I (SR-BI), and the tight junction proteins claudin-1 and occludin.7
To date, the majority of reports have studied HCV replication in hepatocytes or hepatoma-derived cells. However, HCV has been reported to replicate to low levels in nonhepatic cells,9, 10 suggesting that additional cellular reservoirs exist. In this study, we show that human brain microvascular endothelium, the major component of the blood-brain barrier (BBB), expresses all major HCV entry receptors. Furthermore, 2 independently derived brain microvascular endothelial cell lines support HCV entry and replication,11, 12 providing a potential mechanism for HCV to infect the CNS.
Discussion
HCV infection leads to progressive liver disease, which has been associated with extrahepatic syndromes, including CNS abnormalities.3 There is a growing body of literature on mild neurocognitive impairment in chronic HCV infection that is independent of hepatic encephalopathy.27 However, there is a lack of studies to investigate whether cells of the CNS support HCV replication. In this study, we report that all of the essential HCV receptors are expressed on brain microvascular endothelial cells. Indeed, the microvascular endothelia were the only cell type in the brain that expressed all the factors required for HCV entry. To our knowledge, this is the first study to investigate the expression of HCV receptors in the human brain. Microvascular endothelial cells are a major component of the BBB28 and may provide a portal for HCV to infect the CNS.
Quantification of HCV RNA from matched samples of white and grey matter, cerebellum, medulla, liver, and plasma revealed that, in clinical samples with detectable brain HCV, the viral load was 1000- to 10,000-fold lower in brain compared with liver from the same subject. HCV RNA was detected in at least one brain region from 4 of 10 HCV-infected subjects, independent of HIV coinfection status. Although quantities of viral RNA from the brain and liver varied widely between clinical samples, in general a lower viral load was associated with a higher postmortem interval, suggesting RNA degradation in some samples over time. Variation between brain-, plasma-, and liver-derived HCV E1 and 5' untranslated region sequences has previously been reported in this cohort, supporting the hypothesis that HCV replicates and evolves within the brain.29 However, care is needed when interpreting the physiologic relevance of detecting HCV RNA genomes. It is worth noting that 6 HCV-infected subjects had no detectable viral RNA in the brain, despite having comparable levels of viral RNA in the liver and plasma (Supplementary Materials and Methods) to 4 subjects with no detectable HCV RNA in the brain.
There have been significant difficulties in visualizing HCV antigen-e-xpressing hepatocytes in the liver that most likely reflect the low viral burden at a cellular level.30, 31 Our quantification of HCV RNA in brain tissue compared with liver suggests that detecting HCV antigen in the brain will be technically challenging, and current imaging methodologies lack the sensitivity to reliably detect HCV-infected cells in the CNS. Indeed, our attempts to show NS3 or NS5A HCV antigen in brain or liver samples from subjects in this study have failed to provide robust signals (data not shown). Previous studies have reported the presence of HCV RNA in microglia and astrocytes isolated using laser capture microdissection.32, 33 However, our experiments show that astrocytes and microglia lack expression of several receptors required for HCV entry.10
Our studies show that 2 independently derived brain microvascular endothelial cell lines, hCMEC/D3 and HBMEC, support HCVpp entry. Infection was inhibited by antibodies specific for CD81, SR-BI, claudin-1, or E2 glycoprotein, showing a common receptor-dependent entry pathway to that reported for hepatocytes and hepatoma-derived cell lines.34, 35 These observations, along with our recent report that neuroepithelioma cell lines derived from peripheral tumors support efficient HCVpp infection,10 show that viral entry is not restricted to hepatocytes. Importantly, messenger RNA and protein profiling databases show that CD81, SR-BI, claudin-1, and occludin are expressed in epithelial and endothelial cells from various tissues,36, 37 raising the possibility that other cell types may support HCV infection. Our data support the presence of functional entry receptors in BBB endothelial cells but not endothelial cells derived from umbilical vein and liver sinusoids.
Given the permissivity of brain endothelial cells for HCVpp entry, we investigated their ability to support HCVcc replication. HCV-infected hCMEC/D3 cells release low levels of virus that can infect hepatoma cells and showed evidence for a spreading infection that is CD81 dependent. Recent reports highlighting the role of ApoE in HCV assembly8, 24 and the targeting of ApoE-containing nanoparticles across the BBB38, 39 prompted us to investigate a role for ApoE in brain endothelial cell infection. Interestingly, antibodies targeting ApoE effectively neutralized HCV infection of hCMEC/D3 cells, despite modest neutralization of Huh-7 cells, suggesting a greater role for ApoE in virus infection of brain endothelial cells.
The BBB limits the passage of substances from blood to the CNS by the presence of tight junctions between endothelial cells and by receptor-mediated efflux transport systems that restrict the entry of hydrophilic molecules to the brain.28 Multidrug resistance proteins, including P-glycoprotein, restrict the transport of many drugs across the BBB, including antivirals that may contribute to the development of "sanctuary sites," allowing pathogens (eg, HIV-1) to replicate in the brain of drug-treated patients.40 In the present study, HCV replication was inhibited by antiviral agents targeting NS3 protease and NS5B polymerase enzymes in vitro.
Disruption of BBB integrity can lead to an infiltration of pathogens, cytokines, and immune cells to the brain parenchyma as reported for HIV-1 and West Nile viruses.41, 42 hCMEC/D3 infection led to increased HCV RNA and antigen expression over time, with a cytopathic effect that associated with TUNEL staining and increased permeability to the paracellular permeability marker FD-70. These data support a model in which HCV infection may compromise BBB integrity, with implications for brain homeostasis in HCV infection.
In conclusion, we show that brain microvascular endothelium expresses all the major viral receptors required for HCV infection. Two brain endothelial cell lines support HCV entry and replication, in which infection is inhibited by HCV receptor-specific antibodies, interferon, and specific antiviral agents. The observation that HCV-infected hCMEC/D3 cells release low levels of infectious virus and show evidence of apoptosis supports a model in which the BBB may provide an extrahepatic target for infection, and HCV may directly induce neuropathology in vivo.
Results
HCV RNA Load in Brain and Liver Tissue
To quantify HCV RNA levels in the brain and liver of infected subjects, cellular RNA was extracted from human brain (cerebellum, medulla, white and grey matter) and liver from 10 HCV-infected and 3 uninfected subjects as previously described.22 HCV RNA was amplified from the liver sample of all infected subjects tested but not from HCV-seronegative individuals. HCV RNA was detected in brain tissue from 4 of 10 HCV-infected individuals, independent of human immunodeficiency virus (HIV) status (Table 1). In those individuals in whom HCV was detectable in the brain, viral RNA quantities were 1000 to 10,000 times lower than in the matched liver samples (Table 1).
Human Brain Endothelium Express HCV Receptors
To investigate the expression of HCV receptors in the brain, sequential sections from normal and HCV-infected brain samples were stained with antibodies specific for HCV receptors and cell lineage markers: von Willebrand factor (endothelium), glial fibrillary acidic protein, CD68 (macrophages/microglia), and CD163 (perivascular macrophages). Microvascular endothelium costained with endothelial-specific von Willebrand factor and HCV receptors CD81, SR-BI, claudin-1, occludin, and LDL-R (Figure 1 and Supplementary Figure 1). Comparable patterns of viral receptor staining were observed independent of HCV status. CD81, claudin-1, and occludin were also expressed on neurons and CD81 on astrocytes. In contrast, SR-BI expression was restricted to microvascular endothelium (Supplementary Figure 1).
Human Brain Endothelial Cells Support HCV Entry and Replication
Two independently derived brain microvascular endothelial cell lines, hCMEC/D3 and HBMEC,11, 12 express all the HCV entry factors (Figure 2A-C). In contrast, human umbilical vein endothelial cells and liver sinusoidal endothelial cells express low levels of SR-BI and undetectable claudin-1 (Figure 2C), suggesting that expression of the full complement of HCV receptors is specific to brain endothelial cells. Costaining of brain endothelial cells with antibodies specific for CD81, SR-BI, and claudin-1 confirmed expression of all receptors (Supplementary Figure 2).
To ascertain whether viral receptors on brain endothelial cells are functionally active, we studied the ability of HCVpp to infect brain endothelial cells. HCVpp infected hCMEC/D3, HBMEC, primary hepatocytes, and control Huh-7 hepatoma cells, whereas there was no detectable luciferase signal in human umbilical vein endothelial cells and liver sinusoidal endothelial cells (Figure 3A). VSV-Gpp infected all cells with different efficiencies, most likely reflecting cell type-dependent differences in reporter expression. Normalizing HCV infection relative to VSV-G shows comparable HCV entry rates in brain endothelial cells and primary hepatocytes (Figure 3B). Furthermore, HCVpp expressing diverse envelope glycoproteins cloned from several acutely infected subjects infected hCMEC/D3 cells (Figure 3C). HCVpp infection of hCMEC/D3 or HBMEC was inhibited by anti-HCV E2 and patient-derived pooled Ig, confirming glycoprotein-dependent entry (Figure 3D). To investigate the receptor dependency of HCVpp infection of brain endothelial cells, we assessed the ability of monoclonal antibodies specific for viral receptors to neutralize infection. Antibodies specific for CD81, SR-BI, and claudin-1 significantly reduced HCVpp infection of hCMEC/D3 and HBMEC (Figure 3D) but had no effect on VSV-Gpp infection (Supplementary Figure 3), showing a common receptor-dependent pathway of entry in these cell lineages.
Given the permissive nature of brain microvascular endothelial cells for HCV glycoprotein-dependent pseudoparticle infection, we investigated their ability to support replication of 2 chimeric HCVcc JFH-1 viruses expressing genotype 2a strain J619 or genotype 5a strain SA13 structural proteins.18 As controls, we included permissive Huh-7 and nonpermissive U87 cell lines.10 Cells were incubated with increasing concentrations of virus, fixed and infection visualized by staining for viral nonstructural protein NS5A. Both HCV strains infected hCMEC/D3 cells with an approximate 100- to 300-fold reduced titer compared with Huh-7 cells (Figure 4A). HCVcc showed a 100-fold reduced titer in HBMEC compared with hCMEC/D3 cells. Interferon alfa inhibited HCVcc infection of all cell lines (Figure 4A). Unsurprisingly, we failed to detect NS5A in U87 cells (data not shown). To confirm de novo HCV replication and ascertain the sensitivity of endothelial cells to antiviral agents, we compared the ability of several protease and polymerase inhibitors to inhibit HCV replication in hCMEC/D3 and Huh-7 cells. All antiviral agents significantly reduced HCV infection of both cell types (Figure 4B), with the majority of agents showing greater efficacy in hCMEC/D3 cells.
Pretreatment of hCMEC/D3 and Huh-7 cells with antibodies specific for CD81, SR-BI, or claudin-1 significantly reduced HCVcc infection (Figure 4C), confirming our earlier observations with HCVpp. Infectious HCVcc particle assembly is dependent on the lipoprotein synthesis machinery of the host cell leading to the genesis of lipoviral particles.23 Several reports have shown a key role for apolipoprotein E (ApoE) in HCV assembly and entry.8, 24 Because ApoE is known to bind SR-BI and several members of the LDL-R family, we investigated the effect(s) of antibodies targeting ApoE and LDL-R on HCV infection of hCMEC/D3. Anti-ApoE and anti-LDL-R significantly reduced infection of hCMEC/D3 while showing a more modest effect on Huh-7 cells (Figure 4C).
To investigate whether HCV initiates a spreading infection in hCMEC/D3, virus was allowed to enter and infect endothelial or Huh-7 cells for 8 hours, unbound virus was removed by extensive washing, and receptor-specific neutralizing antibodies were added. Virus-infected cells were incubated for 72 hours to allow secondary transmission events to occur and the number of NS5A-expressing infected cells enumerated. Infected foci of NS5A-expressing hCMEC/D3 cells were readily observable, indicating viral spread (Figure 4C). Although receptor-specific antibodies reduced primary infection of hCMEC/D3 cells, antibodies specific for CD81 or ApoE significantly reduced secondary infection, showing a role for cellular CD81 and virus-associated ApoE in viral dissemination between brain endothelial cells (Figure 4D). This ApoE dependency in the endothelial cultures is consistent with endogenous ApoE expression in hCMEC/D3 cells (data not shown).
To confirm a productive infection of brain endothelial cells, HCV SA13/JFH-infected hCMEC/D3 or Huh-7 cells were sequentially harvested to quantify the frequency of NS5A-expressing cells and HCV RNA levels over time. HCV RNA was first detected in hCMEC/D3 cells at 24 hours and levels increased significantly by 48 hours, in parallel with the increasing number of NS5A-expressing cells (Figure 5). There was no detectable viral RNA at 12 hours after infection, showing de novo rounds of viral replication. HCV RNA and NS5A expression in Huh-7 cells increased over time (Figure 5). At 48 hours, the level of HCV RNA in Huh-7 cells was approximately 1000-fold higher than in hCMEC/D3 cells (Figure 5A). However, normalizing HCV RNA to the number of NS5A-positive cells in each culture at 48 hours showed 133 and 1067 copies/infected cell for hCMEC/D3 and Huh-7 cells, respectively, reflecting only a ~10-fold difference in viral genomic burden for the 2 cell types.
miR-122 is a liver-specific microRNA that is required for efficient HCV replication and is considered a therapeutic target for antiviral intervention.25 qRT-PCR confirmed that miR-122 was not detectable in human brain tissue, whereas abundant levels were observed in all liver samples studied. We failed to detect miR-122 expression in hCMEC/D3 cells by qRT-PCR or Northern blot (Supplementary Figure 4A-C). Importantly, transfection of hCMEC/D3 to express functionally active miR-122 RNA duplexes25 failed to promote HCV infection (Supplementary Figure 4D-F), showing that HCV replication in hCMEC/D3 cells is miR-122 independent.
Brain Endothelial Cells Release Infectious Virus
To ascertain whether brain endothelial cells can release virus that is infectious for hepatocytes, hCMEC/D3, Huh-7, or nonpermissive U87 cells were infected with HCVcc SA13/JFH for 12 hours at a comparable multiplicity of infection and unbound virus was removed by washing. Virus-infected cells were incubated at 37°C for 72 hours, extracellular medium was collected, and cells were fixed and stained for NS5A. Levels of infectious virus in the medium were determined by inoculating na•ve Huh-7.5 cells. Similar numbers of HCV-infected hCMEC/D3 and Huh-7 cells were observed; however, the level of infectious virus released from hCMEC/D3 cells over an 8-hour period was 68 focus-forming units, compared with 1680 focus-forming units released from Huh-7 cells (Supplementary Figure 5). We attempted to inoculate na•ve hCMEC/D3 cells with viral supernatant from HCVcc-infected hCMEC/D3 cells and failed to detect infection, most likely because of the low levels of virus released from infected hCMEC/D3 cultures (data not shown). To ascertain whether the low levels of infectious virus in the hCMEC/D3 culture medium could be attributed to a persisting viral inoculum, we titered the extracellular media collected from virus-inoculated U87 cells and failed to detect infectious virus. Furthermore, medium collected 12 hours after virus inoculation contained no detectable infectious virus. In summary, these data show that brain endothelial cells release low levels of HCV that are infectious for hepatoma cells.
HCV Increases Endothelial Cell Permeability
To investigate whether HCV infection affects hCMEC/D3 paracellular permeability, confluent cells were allowed to polarize and 70-kilodalton fluorescein isothiocyanate/dextran flux was measured. Human tumor necrosis factor α/interferon gamma treatment or HCV infection significantly increased hCMEC/D3 paracellular permeability (Figure 6A), showing that HCV can disrupt endothelial cell integrity. Neutralization of HCV infection with pooled anti-HCV patient Ig restored hCMEC/D3 impermeability, showing a direct effect of infection (Figure 6A). We noted that HCV-infected hCMEC/D3 cells showed evidence of cytopathicity in association with NS5A expression. To ascertain whether HCV induced apoptosis, we stained infected hCMEC/D3 and Huh-7 cells for DNA strand breaks using TUNEL. A low frequency of HCV-infected Huh-7 cells stained positive for both TUNEL and NS5A.26 In contrast, all the NS5A-positive endothelial cells were TUNEL positive, showing a direct effect of infection on brain endothelial cell apoptosis (Figure 6B).
Materials and Methods
Cells, Reagents, and Clinical Material
Huh-7 and 293T cells were provided by C. Rice (Rockefeller University, New York, NY) and U87 cells by American Type Culture Collection (Manassas, VA). All cells were maintained in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum, 1% nonessential amino acids/1% penicillin/streptomycin (Invitrogen). hCMEC/D3 cells were maintained in complete EGM-2 medium (Lonza, United Kingdom).12 HBMEC cells were maintained in RPMI supplemented with 10% fetal bovine serum/10% NuSerum and 30 μg/mL Endothelial Cell Growth Supplement (BD Biosciences) as well as 1% nonessential amino acids/1% penicillin/streptomycin (Invitrogen). Human umbilical vein endothelial cells and liver sinusoidal endothelial cells were isolated as previously described.13 Clinical material is further described in Supplementary Materials and Methods.
The primary antibodies were anti-NS5A 9E10 (C. Rice, Rockefeller University), anti-CD81 (2.131),14 anti-SR-BI (V. Flores, Pfizer), anti-claudin-1 (Abnova and R&D), anti-claudin-1 polyclonal sera,15 anti-occludin (Invitrogen), anti-ZO-1 (Invitrogen), anti-LDL-R (Progen), anti-apolipoprotein E (mAb23),16 anti-von Willebrand factor (Dako), anti-glial fibrillary acidic protein (Dako), anti-CD63 (Dako), anti-CD163 (Novocastra), and anti-E2 (9/27, 11/27, and 3/11).17 Immunoglobulin (Ig) from healthy volunteers and chronically HCV-infected donors was purified by protein G affinity chromatography. Fluorescent secondary antibodies Alexa Fluor 488 and 594 anti-mouse, anti-human, anti-rat, and anti-rabbit IgG were obtained from Invitrogen.
Flow Cytometric Analysis of Receptor Expression
For CD81, SR-BI, claudin-1, ZO-1, and LDL-R staining, cells were incubated with monoclonal antibodies at 2 μg/mL in phosphate-buffered saline containing 1% bovine serum albumin/0.01% sodium azide for 20 minutes at 37°C. For occludin staining, cells were fixed with 1% paraformaldehyde and permeabilized with phosphate-buffered saline/1% bovine serum albumin/1% saponin. Bound antibodies were detected with Alexa 488 secondary antibodies and quantified by flow cytometry using a FACSCalibur (BD Biosciences) and FlowJo software (TreeStar, Ashland, OR).
Laser Scanning Confocal Microscopy
Sequential 5-μm sections of formalin-fixed, paraffin-embedded normal brain tissue from 5 subjects were dewaxed and rehydrated and microwave antigen retrieval was performed in EDTA buffer. Cell lines were plated on collagen-coated coverslips (Fisher Scientific, United Kingdom) and 24 hours later fixed with ice-cold methanol (claudin-1, occludin, ZO-1, LDL-R) or 3% paraformaldehyde (CD81). After incubation with primary antibodies (2 μg/mL), cells were incubated with Alexa 488 secondary antibodies, counterstained with 4',6-diamidino-2-phenylindole, and viewed by laser scanning confocal microscopy on a Zeiss MetaHead microscope with a 100x oil immersion objective.
HCVpp and HCVcc Genesis and Infection
Luciferase reporter pseudoparticles expressing a panel of HCV envelope glycoproteins (HCVpp), vesicular stomatitis virus G glycoprotein (VSV-Gpp), or a no-envelope control were generated as previously reported.17 Virus-containing medium was added to cells in 96-well plates seeded at 7.5 x 103 cells/cm2 and incubated for 72 hours. Cells were lysed, and luciferase activity was measured (Lumat LB9507 luminometer). Infectivity is expressed as relative light units, where the no-envelope signal is subtracted from HCVpp or VSV-Gpp signals.
Plasmids encoding chimeric SA13/JFH18 or J6/JFH19 were used to generate RNA as previously described.19 Briefly, RNA was electroporated into Huh-7.5 cells, and supernatants were collected at 72 and 96 hours and stored at -80°C. Infected cells were fixed with ice-cold methanol and stained for NS5A with monoclonal antibody 9E10 and isotype-matched Alexa 488-conjugated anti-mouse IgG2a. NS5A-positive foci were enumerated, and infectivity was expressed as focus-forming units per milliliter.
Neutralization of HCV Infection
Cells were seeded in 96-well plates at 7.5 x 103 cells/cm2. After 24 hours, cells were incubated with anti-receptor or irrelevant IgG control monoclonal antibodies at 10 μg/mL for 1 hour, and HCVcc or HCVpp was added and incubated for 72 hours. Anti-E2 monoclonal antibodies or HCV-positive IgG were incubated with virus for 1 hour before infecting target cells. Alternatively, cells were incubated with virus for 8 hours, unbound virus was removed by washing, and cells were incubated with neutralizing antibodies. For HCVpp, cells were lysed and luciferase activity was measured. For HCVcc, cells were fixed with ice-cold methanol and stained for NS5A. The percent neutralization was calculated relative to the irrelevant IgG.
Real-Time Reverse-Transcription Polymerase Chain Reaction
Purified cellular or tissue RNA samples were amplified for HCV RNA (Primer Design Ltd) in a quantitative reverse-transcription polymerase chain reaction (qRT-PCR) as per the manufacturer's guidelines (CellsDirect Kit; Invitrogen) and fluorescence was monitored in an MxPro-3000 PCR machine (Stratagene). Comparison of a panel of housekeeping genes in brain and liver RNA confirmed GAPDH as a stably expressed reference gene. Hence, GAPDH was included as an endogenous control for amplification efficiency and RNA quantification. The assay cutoff was 100 copies.
Permeability and Apoptosis Assays
hCMEC/D3 cells were seeded on collagen I-coated Transwell filters (0.4 μm; pore size, 1.1 cm2; Sigma-Aldrich) and cultured as described.20 Cells were infected with HCVcc, with or without prior incubation with anti-HCV IgG for 48 hours, or a combination of tumor necrosis factor α and interferon gamma for 24 hours before measuring paracellular permeability by using the clearance method.21 hCMEC/D3 and Huh-7 cells were seeded on collagen-coated coverslips and infected with J6/JFH or SA13/JFH. After 72 hours, cells were fixed and terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick-end labeling (TUNEL) staining was performed as per the manufacturer's instructions (Invitrogen).
Statistical Analysis
Statistical analyses were performed using Student t test in Prism 4.0 (GraphPad, La Jolla, CA), with P < .05 considered significant.
|
|
| |
| |
|
|
|