| |
Achillion Announces Positive SVR4 Results From Phase 2 Study of Sovaprevir (Formerly ACH-1625) and Advancement of ACH-3102
|
| |
| |
Sovaprevir (formerly ACH-1625) achieves SVR4 of 85-100% of genotype 1 treatment naive patients treated with sovaprevir for 12 weeks followed by an additional 12 weeks of pegylated-interferon and ribavirin
Enrollment of HCV-infected patients initiated in Phase 1 trial of ACH-3102, second generation pan-genotypic NS5A inhibitor
Conference call tomorrow August 8, 2012 at 10:00 a.m. EDT
NEW HAVEN, Conn., Aug. 7, 2012 (GLOBE NEWSWIRE) -- Achillion Pharmaceuticals, Inc. (Nasdaq:ACHN) today announced sustained viral response (SVR4) results of 85 to 100 percent from an ongoing multi-dose Phase 2 trial evaluating 12 weeks of dosing with sovaprevir (formerly ACH-1625), a once-daily protease inhibitor, in combination with pegylated interferon plus ribavirin (P/R) followed by an additional 12 weeks of P/R. In addition, the Company announced today that ACH-3102, a second-generation pan-genotypic NS5A inhibitor, has been safe and well tolerated by healthy volunteers in both single and 14-day multiple ascending dose groups. Further, enrollment of patients in a Phase 1 proof-of-concept clinical trial to evaluate the safety and efficacy of ACH-3102 in patients with genotype 1 HCV has been initiated.
"We are very pleased to reach these important milestones in our HCV portfolio including positive SVR4 results with sovaprevir and the advancement of ACH-3102, our second-generation pan-genotypic NS5A inhibitor, through Phase 1 dose-escalation," commented Milind S. Deshpande, Ph.D., President of Research and Development and Chief Scientific Officer. "The safety and efficacy seen with sovaprevir across dose groups provides us with confidence that this next-generation protease inhibitor will play an important role in an all-oral treatment for HCV. The safety and tolerability seen to date with ACH-3102, for which we expect to report proof-of-concept next month, combined with the compound's in vitro profile, lead us to we believe we have an in-house portfolio of optimized compounds that can successfully create an all-oral, interferon-free regimen for the treatment of genotype 1 HCV. We look forward to beginning combination studies with sovaprevir and ACH-3102 by the end of the year."
Sovaprevir: Updated Phase 2 results including SVR4
In June 2011, Achillion initiated a randomized Phase 2 trial evaluating three doses (200 mg, 400 mg, or 800 mg) of sovaprevir given once daily in combination with pegylated interferon plus ribavirin (P/R) for 12 weeks followed by an additional 12 or 36 weeks of P/R, for the treatment of genotype 1 HCV.
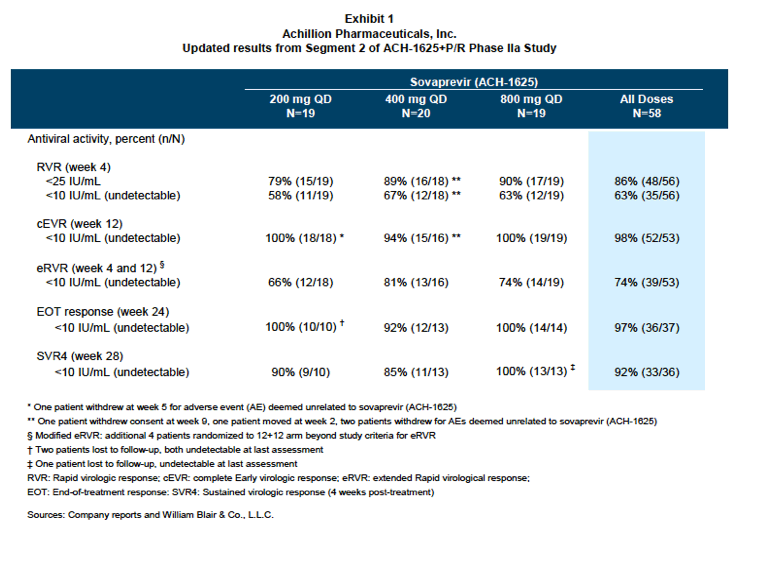
As previously reported in April 2012, of the 58 patients enrolled in this study, the majority had HCV genotype 1a (n=35 (60%)), with remaining patients having HCV genotype 1b (n=20) or genotype 1 (n=3). Approximately 71% of the patients were IL28B genotype CT/TT, the more difficult to treat mutation, 64% were male and 17% were African American. The complete early virologic responses (cEVR) across the 200 mg, 400 mg, and 800 mg sovaprevir dose groups were 100%, 94% and 100%, respectively.
Today, the Company reported SVR4 rates of 90%; 85%; and 100% in the 200 mg, 400 mg, and 800 mg dose groups, respectively, after 24 weeks of therapy consisting of 12 weeks of sovaprevir and P/R followed by additional 12 weeks of P/R. In all, 39 patients were assigned to receive an additional 12 weeks of P/R therapy with the remaining 14 patients assigned to receive an additional 36 weeks of P/R.

As previously reported, sovaprevir was generally well tolerated across all dose groups. Adverse events (AEs) in patients receiving sovaprevir were classified as mild to moderate and were transient. The most common AEs were consistent with P/R treatment.
Additional clinical trial results, including SVR4 and SVR12 for all patients treated with sovaprevir followed by an additional 12 weeks or 36 weeks of P/R, are expected be reported during the first quarter of 2013.
ACH-3102: Phase 1 trial in Healthy Volunteers and HCV-infected patients
In May 2012, Achillion initiated a Phase 1 clinical trial evaluating the safety and tolerability of single and multiple ascending doses of ACH-3102, a once daily, second-generation, pan-genotypic NS5A inhibitor, in healthy volunteers.
To date, 42 healthy volunteers have received a single dose of ACH-3102, ranging from 25 mg to 1,000 mg. An additional 24 healthy volunteers have received 14 days of once daily ACH-3102, with doses ranging from 25 mg to 75 mg. Preliminary data from both the single and multiple ascending dose groups demonstrated that ACH-3102 was well tolerated at all doses evaluated. There were no serious adverse events, no clinically significant changes in vital signs, electrocardiograms (ECGs), or laboratory evaluations. All reported adverse events were classified as mild or moderate, and were transient in nature.
Achillion announced today the initiation of enrollment of patients with genotype 1 HCV into a Phase 1 study to evaluate the safety, tolerability and antiviral activity of ACH-3102. The trial will initially evaluate the safety and antiviral activity of a single dose of ACH-3102. Initial results are expected to be reported during the third quarter of 2012.
from William Blair stock market Analysts
Y. Katherine Xu, Ph.D.
Key conclusions and thoughts on the data include:
The 85%100% SVR4 achieved in the eRVR responders by sovaprevir+P/R is inline with expectations for a potent PI. We note that such data is comparable to those observed with compounds in the PI class and from other classes involved with similar designs.
Is there a dose response for sovaprevir? At the lower doses of 200 mg and 400 mg, relapses were observed, while at the highest dose of 800 mg, the SVR4 was 100%. As the sample size of the study is small, it is impossible to definitively conclude whether there is a dose response or not. Pharmocokinetic (PK) analyses indicate that even at the lowest dose of 200 mg, the trough plasma concentration achieved is far higher than the EC50 to inhibit viral replication. Further, the RVR, EVR, eRVR, and EOT rates are not different among the three dose groups, as illustrated in exhibit 1. These observations support the argument that the three doses are likely equivalent.
Is the sovaprevir safety dataset sufficient leading into the combo study? To date, only 53 patients have been treated with sovaprevir for 12 weeks, and for a HCV drug candidate to be viable, 12-week safety is a critical bar to clear. We have seen strategies from other companies to combine their novel candidate with multiple other direct antiviral agents (or P/R or R alone) that are well characterized on the safety side to build up a safety database consisting of 200-300 patients. Achillion intends to go into a proprietary 12-week sovaprevir+ACH-3102 combo study by year-end, which raises the question whether the current sovaprevir safety database is sufficient; a larger safety database would certainly provide higher confidence and more comfort.
Sovaprevir dose selection going forward: the lower doses of 200 mg and 400 mg are likely to be selected. As discussed above, the efficacy signals across the dose groups appear to be similar, and the PK data demonstrates sufficient plasma trough levels even at the lowest dose; we note that sufficiently high trough levels are important for preventing viral breakthrough. On the safety side, two ALT elevations occurred in the 400 mg dose group, two occurred in the 800 mg dose group, and none were reported from the 200 mg dose group (although 3 of the 4 cases were deemed "not related to sovaprevir" and there is no judgment call on the fourth case). Management indicated that the 200mg QD and 400 mg QD doses would be selected to go forward, which represents better efficacy/risk ratio.
Sovaprevir+P/R demonstrates initial SVR4 of 85%100% in patients who achieved an eRVR, in line with our expectations for a nextgeneration,
potent protease inhibitor (exhibit 1). In the Phase IIa study, patients were first treated with 12 weeks of sovaprevir+P/R; a total of 39 out of 53 patients were considered achieving eRVR (early rapid virologic response, viral negative at both weeks 4 and 12) and assigned to receive an additional 12 weeks of P/R (12+12 arm), while the remaining 14 were allocated to receive an additional 36 weeks of P/R (12+36 arm). For the eRVR patients on the 12+12 arm, 9 of 10 (90%), 11 of 13 (85%), and 13 of 13 (100%) achieved an SVR4 in the sovaprevir 200 mg, 400 mg, and 800 mg dose groups, respectively. The 12+36 arm is still ongoing, with SVR data expected during first quarter 2013.
Two relapses and one viral breakthrough on P/R. Management noted that two patients relapsed, one from the 200 mg cohort and the other from the 400 mg cohort. Both patients were of GT1a and IL28B TT genotype status. Achillion also reported that there was one patient who experienced a viral rebound in the 400 mg arm while on P/R alone; this patient was of GT1b with IL28B CC status. All three patients had high-viral load at baseline, ranging from 5.94 logs to 6.24 logs.
No additional safety signals reported. As previously reported, sovaprevir was generally well tolerated across all dose groups; the most-common adverse events (AEs) were consistent with the treatment of P/R alone. The most notable AEs were the transient bilirubin increases that start at week 2 and resolve at weeks 4-5. Further, at this year's EASL (European Association for the Study of the Liver) conference (April 18-22, Barcelona), final data from all 58 patients from the 12-week study revealed four cases of liver enzyme elevations. While the first three patients had grade 3 or 4 ALT elevations, the AEs were deemed by the principal investigators as "not related to study drug" because of protocol violations or incomplete suppression of the HCV virus. The fourth case is milder at grade 2, and there is no formal judgment from the principal investigator as to whether this event was related to the study drug or not.
Review of Segment 2 from the ACH1625+P/R Phase II study. Segment 2 of the study enrolled and randomized 58 treatment-na•ve GT1 patients to sovaprevir at 200 mg QD, 400 mg QD or 800 mg QD, in combination with P/R. Sixtysix percent of patients were of GT1a, 33% were of the IL28B CC genotype, 74% were male, and 14% were African American. The 48-week P/R control arm from segment 1 of the Phase IIa study will serve as the control for segment 2. In segment 1, Achillion enrolled a similar difficult-to-treat GT1 population (73% of patients were of GT1a, and only 17% were of the IL-28B CC genotype) and generated a RVR of 75%-81%, among the highest in the class.
ACH3102 safe in 14-day Phase I study in healthy volunteers; initial viral kinetic data in HCV patients expected during the third quarter. In May, Achillion initiated the placebo-controlled Phase I study to evaluate safety, tolerability, and pharmacokinetic profile of ACH-3102 in healthy volunteers. To date, ACH-3102 has been dosed in 64 healthy volunteers; 42 in the single-ascending dose (SAD) segment, and 24 in the multiple-ascending dose (MAD) segment. In the MAD segment, volunteers received 14 days of once daily (QD) ACH-3102, with doses ranging from 25 mg to 75 mg. All reported AEs were classified as mild or moderate, were transient in nature, and no serious AEs or laboratory abnormalities were observed. In the recently initiated third segment of the Phase I study, Achillion will evaluate the safety, tolerability, and antiviral activity of single-dose administration of ACH-3102, exploring three doses of ACH-3102 (50 mg, 150 mg, and 300 mg) in GT1 patients. Management plans to report initial antiviral activity during third quarter 2012.
Sovaprevir +ACH3102 DDI study to be conducted during the third quarter, leading to a 12-week combination study to be initiated by yearend 2012, as previously guided; topline results from the combo study expected in first quarter 2013. Achillion reiterated its plans to initiate a two-week drug-drug interaction (DDI) study of ACH-1625+ACH-3102 in the third quarter to support a subsequent 12-week, all-oral combination study of sovaprevir +ACH-3102, with or without R, in GT1 HCV patients during fourth quarter 2012. Achillion noted that it may decide to include a 24-week arm in the study. We anticipate initial results from the study in first quarter 2013.
|
|
| |
| |
|
|
|