 |
|
|
| |
Targeting lysyl oxidase like 2 (LOXL2) inhibits
collagen cross-linking and accelerates reversal of
pre-established liver fibrosis
|
| |
| |
Reported by Jules Levin
AASLD 2013 Nov 1-4 Wash DC
Naoki Ikenaga1, Shuhei Yoshida1, Susan B. Liu1, Jeanhee Chung1, Deanna Sverdlov1,
Derek Marshall2, Vivian Barry2, Victoria Smith2, Maria Kovalenko2, Satyajit Karnik2,
Nezam H. Afdhal1, Yury Popov1
1Division of Gastroenterology and Hepatology
Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA
2Gilead Sciences, Foster City, CA
AASLD: Selective inhibition of lysyl oxidase like 2 (LOXL2) using a therapeutic monoclonal antibody suppresses the progression of biliary fibrosis in novel PSC-like mouse model - (11/13/13)
PROGRAM ABSTRACT:
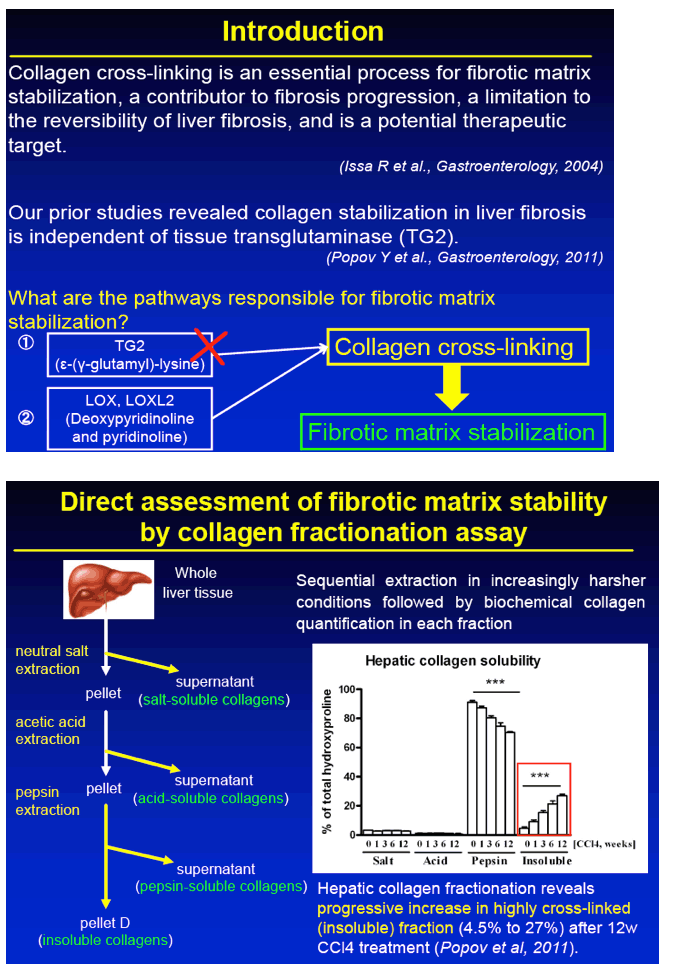
BACKGROUND/AIMS: Emerging evidence suggests that pre-existing cirrhosis caused by chronic hepatitis C confers long-term risk of liver cancer even after the virus has been successfully eliminated. We previously showed that transglutaminase-independent collagen cross-linking retards liver cirrhosis reversal. In this study, we test whether collagen cross-linking can be successfully inhibited to reverse advanced fibrosis in vivo utilizing an antibody directed against LOXL2, an inducible protein with lysyl oxidase activity in mouse models.
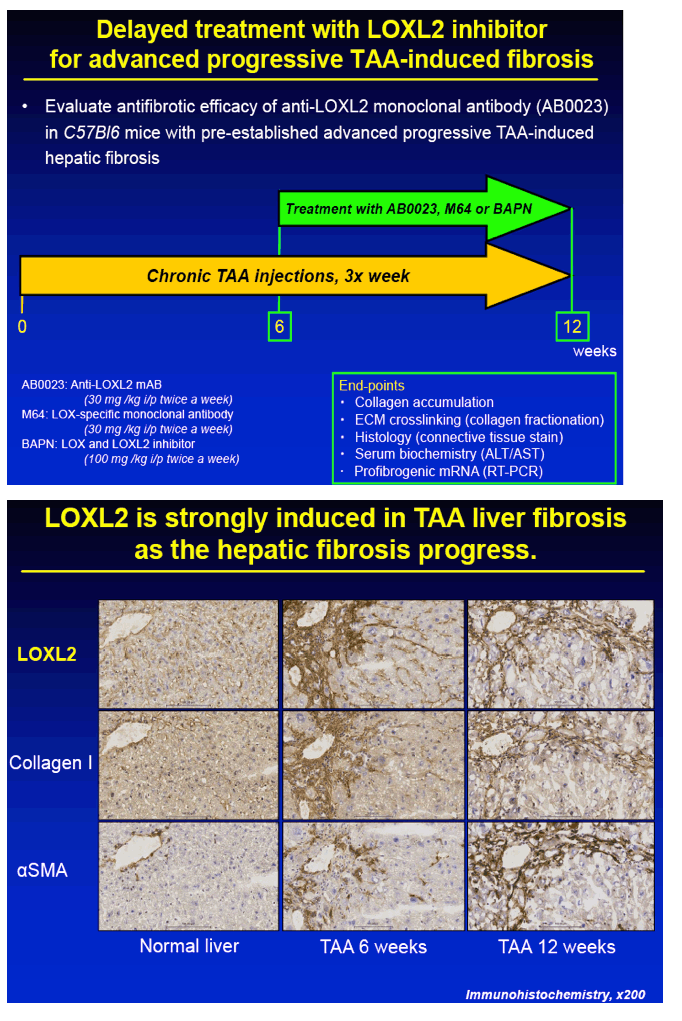
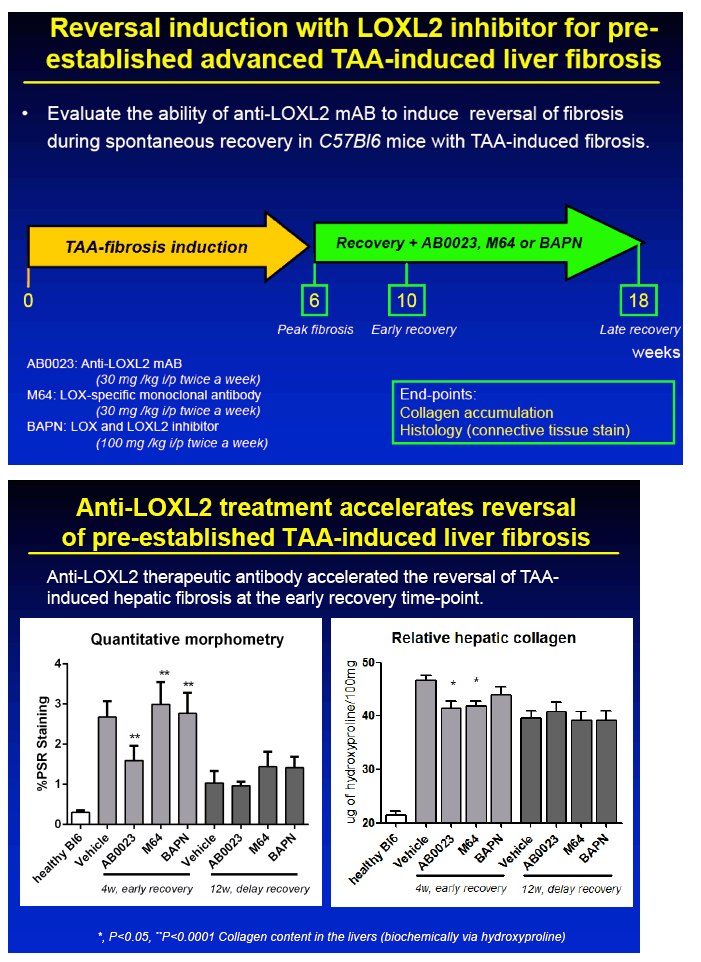
METHODS: Advanced liver fibrosis was induced in C57Bl/6 mice by repeated injections of thioacetamide (TAA). Novel anti-LOXL2 therapeutic antibody (AB0023mAB, 30mg/kg) or control antibody (M64, 30mg/kg) was administered i.p. twice a week (n=10-16 per group) during fibrosis progression (delayed treatment, from week 6 to 12 of TAA) or during fibrosis reversal (recovery, 1 to 12 weeks after TAA). Collagen cross-linking was assessed ex vivo using a step-wise collagen extraction/fractionation method.
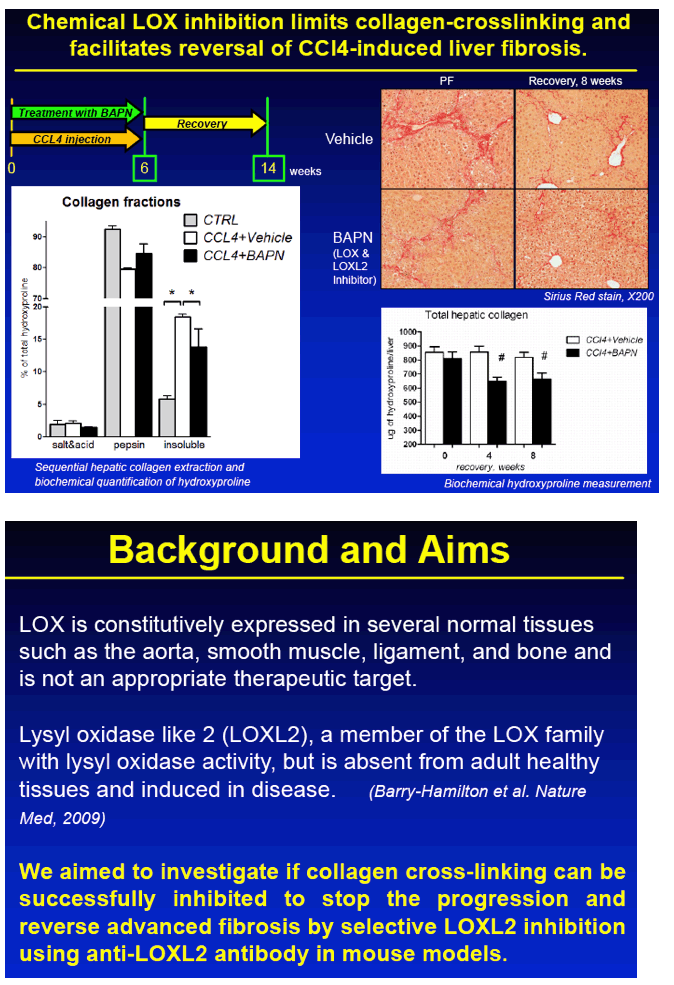
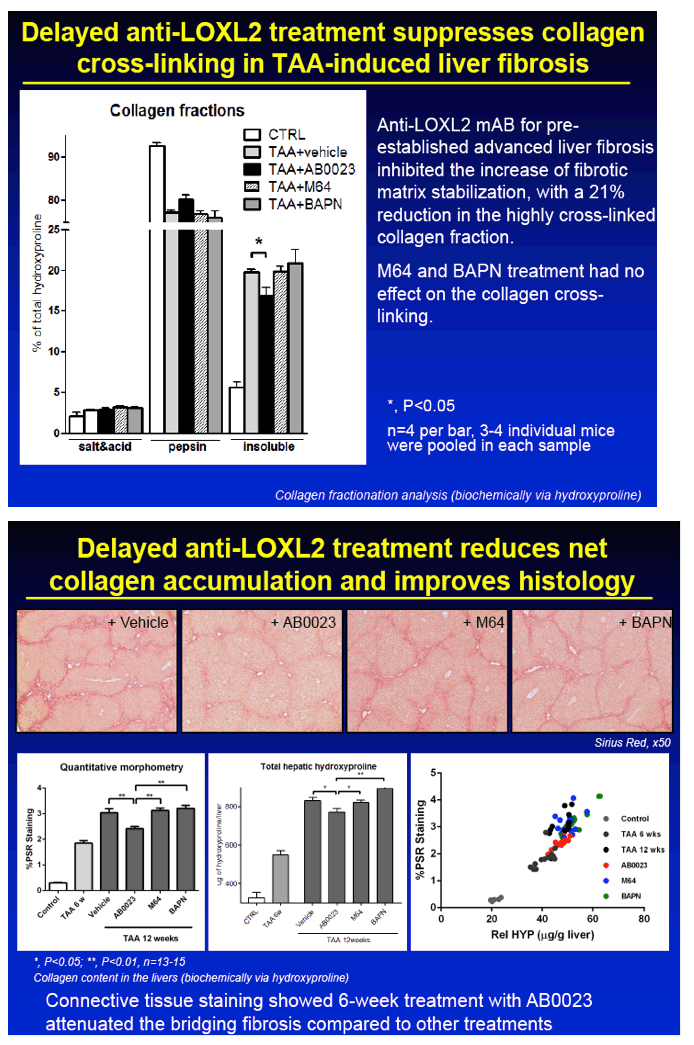
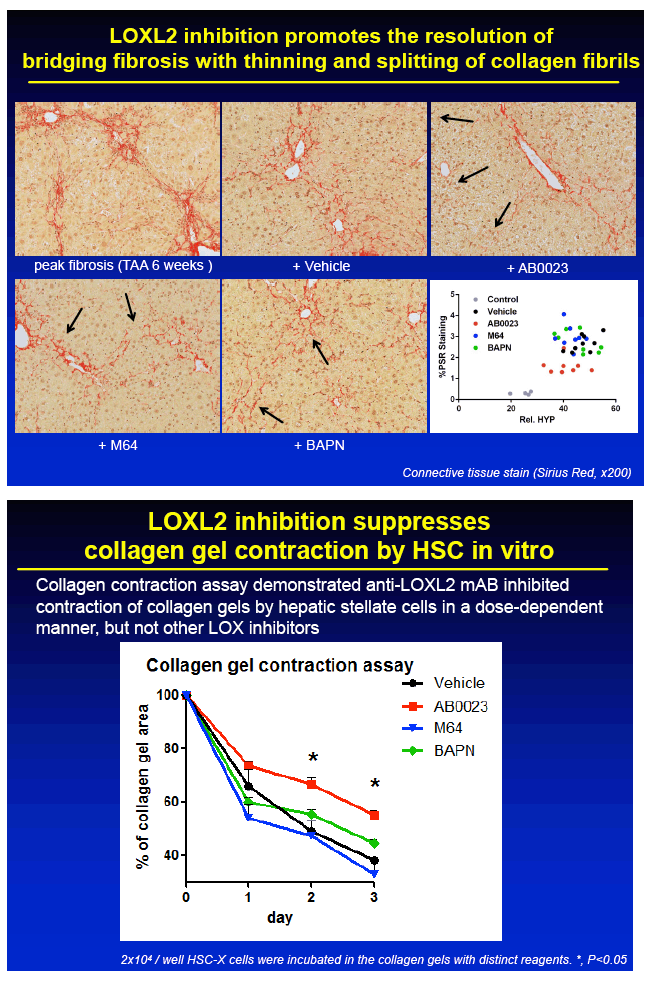
RESULTS: Immunohistochemical analysis revealed that LOXL2 was virtually absent from healthy liver, but was strongly induced in TAA-induced fibrotic liver, with predominant localization within fibrotic septa. Delayed anti-LOXL2 treatment of pre-established, advanced liver fibrosis (week 6 through 12 of TAA) inhibited fibrotic matrix stabilization, with a 30% reduction in the highly cross-linked collagen fraction. Histological signs of bridging fibrosis improved, with a 25% decrease in net collagen deposition in LOXL2-treated group as assessed biochemically via hydroxyproline (p=0.025). When LOXL2 was inhibited during fibrosis recovery, profound acceleration of remodeling of fibrotic septa was observed, with thinning and splitting of collagen fibrils histologically, and a 36% decrease in hepatic collagen levels (p=0.021) peaking at the early recovery time-point (4 weeks). In contrast, no significant effect on collagen cross-linking, fibrosis progression, or reversal was detected using histological or biochemical methods in control antibody -treated mice.
CONCLUSIONS: 1) Antibody-mediated LOXL2 inhibition effectively suppressed collagen cross-linking during experimental liver fibrosis progression in vivo. 2) LOXL2 inhibition rapidly and potently accelerated hepatic fibrosis resolution in the recovery model from TAA-injury. 3) Feasibility of antibody targeting of LOXL2 to prevent and reverse liver cirrhosis should be evaluated in future clinical trials.

|
| |
|
|
|
|
|