| |
Faldaprevir and Deleobuvir for HCV Genotype 1 Infection
|
| |
| |
Download the PDF
Download the PDF
The New England Journal of Medicine
august 15, 2013
Stefan Zeuzem, M.D., Vincent Soriano, M.D., Ph.D., Tarik Asselah, M.D., Ph.D., Jean-Pierre Bronowicki, M.D., Ph.D., Ansgar W. Lohse, M.D., Beat Mullhaupt, M.D., Marcus Schuchmann, M.D., Marc Bourliere, M.D., Maria Buti, M.D., Stuart K. Roberts, M.D., Ed J. Gane, M.D., Jerry O. Stern, M.D., Richard Vinisko, M.A., George Kukolj, Ph.D., John-Paul Gallivan, Ph.D., Wulf-Otto Bocher, M.D., and Federico J. Mensa, M.D.
"Rates of a sustained virologic response 12 weeks after the completion of therapy were 56 to 85% among patients with genotype 1b infection versus 11 to 47% among patients with genotype 1a infection and 58 to 84% among patients with IL28B CC versus 33 to 64% with non-CC genotypes. Rash, photosensitivity, nausea, vomiting, and diarrhea were the most common adverse events."
Background
Interferon-free regimens would be a major advance in the treatment of patients with chronic hepatitis C virus (HCV) infection.
Methods
In this phase 2b, randomized, open-label trial of faldaprevir (a protease inhibitor) and deleobuvir (a nonnucleoside polymerase inhibitor), we randomly assigned 362 previously untreated patients with HCV genotype 1 infection to one of five groups: faldaprevir at a dose of 120 mg once daily and deleobuvir at a dose of 600 mg three times daily, plus ribavirin, for 16, 28, or 40 weeks (TID16W, TID28W, or TID40W, respectively); faldaprevir at a dose of 120 mg once daily and deleobuvir at a dose of 600 mg twice daily, plus ribavirin, for 28 weeks (BID28W); or faldaprevir at a dose of 120 mg once daily and deleobuvir at a dose of 600 mg three times daily, without ribavirin, for 28 weeks (TID28W-NR). The primary end point was a sustained virologic response 12 weeks after the completion of therapy.
Results
The primary end point was met in 59% of patients in the TID16W group, 59% of patients in the TID28W group, 52% of patients in the TID40W group, 69% of patients in the BID28W group, and 39% of patients in the TID28W-NR group. The sustained virologic response 12 weeks after the completion of therapy did not differ significantly according to treatment duration or dosage among ribavirin-containing regimens. This response was significantly higher with TID28W than with TID28W-NR (P=0.03). Rates of a sustained virologic response 12 weeks after the completion of therapy were 56 to 85% among patients with genotype 1b infection versus 11 to 47% among patients with genotype 1a infection and 58 to 84% among patients with IL28B CC versus 33 to 64% with non-CC genotypes. Rash, photosensitivity, nausea, vomiting, and diarrhea were the most common adverse events.
Conclusions
The rate of a sustained virologic response 12 weeks after the completion of therapy was 52 to 69% among patients who received interferon-free treatment with faldaprevir in combination with deleobuvir plus ribavirin. (Funded by Boehringer Ingelheim; SOUND-C2 ClinicalTrials.gov number, NCT01132313.)
The introduction of the direct-acting antiviral agents telaprevir and boceprevir (nonstructural protein 3/4A [NS3/4A] protease inhibitors) was a major advance in the management of chronic infection with hepatitis C virus (HCV) genotype 1, the most prevalent and difficult-to-cure genotype.1 However, these drugs are used in combination with pegylated interferon alfa and ribavirin, which are associated with a high rate of side effects and discontinuation.2 In addition, many patients cannot receive pegylated interferon because of contraindications.3
Host genetic factors are known to influence the response to treatment with pegylated interferon and ribavirin in patients infected with HCV genotype 1. Single-nucleotide polymorphisms in the promoter region of the IL28B gene result in three genotypes: CC, CT, and TT (rs12879860), and patients with the CT or TT genotype have a reduced response to interferon-based therapies.4 The influence of this host factor on a sustained virologic response in patients treated with interferon-free regimens is unknown.
Monotherapy with direct-acting antiviral agents is associated with rapid selection of viral variants that are resistant to the antiviral compound.4-7However, combinations of potent direct-acting antiviral agents targeting different stages of the HCV life cycle offer the possibility of interferon-free treatment. The results of phase 1 and 2 studies involving patients with HCV genotype 1 infection have been encouraging and provide support for further development of combination therapies.8-13 In the phase 1b Safety and Antiviral Effect of Oral Combinations without Interferon in Patients Diagnosed with Hepatitis C (SOUND-C1) study, patients who had not previously been treated for HCV infection received 4 weeks of treatment with faldaprevir (an NS3/4A protease inhibitor) and deleobuvir (formerly known as BI207127; a nonnucleoside inhibitor of nonstructural protein 5B [NS5B] polymerase) plus ribavirin. At week 4, 100% and 73% of patients who received faldaprevir plus ribavirin with 600 mg or 400 mg of deleobuvir, respectively, had HCV RNA levels that were lower than 25 IU per milliliter.12 Here we describe the phase 2b SOUND-C2 study, which assessed the efficacy and safety of the interferon-free combination of faldaprevir plus deleobuvir, with or without ribavirin, for 16, 28, or 40 weeks.
Methods
Patients
Patients were enrolled at 48 sites in Europe, Australia, and New Zealand. Eligible patients were 18 to 75 years of age, had chronic HCV genotype 1 infection (HCV RNA level ≥10,000 IU per milliliter), had compensated liver disease, and had not received treatment. Patients could have cirrhosis, which was determined either by means of biopsy (Metavir stage F4 on a scale from F0 to F4, with higher stages indicating a greater degree of fibrosis) or by means of transient elastography. The study protocol is available with the full text of this article at NEJM.org.
Study Design
The trial was a multicenter, randomized, open-label, phase 2b study. Although no formal hypotheses were stated, the study was designed to investigate the effect of treatment duration, deleobuvir dosage, and the absence or presence of ribavirin in treatment regimens on the virologic response. Patients were stratified at randomization according to viral subtype (1a or 1b, which was determined by means of the Trugene HCV genotyping assay [Bayer] or the Versant HCV Genotype 2.0 Assay [Siemens], if the Trugene result was inconclusive) and according to the IL28B(rs12979860) genotype (either CC or non-CC, which was determined by means of polymerase-chain-reaction TaqMan allelic discrimination assays14 [Applied Biosystems]). With the use of an interactive voice-response system, the patients were assigned in a 1:1:1:1:1 ratio to one of five treatment groups: faldaprevir at a dose of 120 mg once daily and deleobuvir at a dose of 600 mg three times daily, plus ribavirin, for 16 weeks (the TID16W group), 28 weeks (TID28W), or 40 weeks (TID40W); faldaprevir at a dose of 120 mg once daily and deleobuvir at a dose of 600 mg twice daily, plus ribavirin, for 28 weeks (BID28W); and faldaprevir at a dose of 120 mg once daily and deleobuvir at a dose of 600 mg three times daily, without ribavirin, for 28 weeks (TID28W-NR).
For the first dose of the study drugs, patients received a 1200-mg dose of deleobuvir (i.e., an additional 600-mg dose) and a 240-mg dose of faldaprevir (i.e., an additional 120-mg dose). The ribavirin dose was 1000 mg per day (for patients with a body weight of <75 kg) or 1200 mg per day (for those with a body weight ≥75 kg). Stepwise temporary reductions of the ribavirin dose and the use of erythropoietin were permitted to manage anemia.
Patients who had virologic breakthrough or a detectable level of HCV RNA at weeks 6 and 8 were switched to pegylated interferon plus ribavirin and were considered to have had treatment failure. Breakthrough was defined as a confirmed increase in the HCV RNA level in two consecutive measurements to 25 IU per milliliter or higher for patients with HCV RNA levels that were previously lower than 25 IU per milliliter or 1 log10 IU per milliliter or higher for those with HCV RNA levels that were previously 25 IU per milliliter or higher. Relapse was defined as an HCV RNA level that was higher than 25 IU per milliliter after an undetectable level of HCV RNA at the end of planned treatment.
In a protocol amendment that was effective as of December 2011 (2 months before the internal database was unblinded), the definition of a sustained virologic response was changed from an undetectable level of HCV RNA 24 weeks after completion of therapy to an undetectable level of HCV RNA 12 weeks after completion of therapy. This change was made on the basis of a meta-analysis of HCV clinical trials showing that a sustained virologic response 12 weeks after the completion of therapy had a positive predictive value of 98% for a sustained virologic response 24 weeks after the completion of therapy.15 Other protocol amendments are described in the study protocol.
Study Oversight
The study was approved by the ethics committee at each participating site and was carried out in accordance with the Declaration of Helsinki and the International Conference on Harmonisation Guidelines for Good Clinical Practice. All patients provided written informed consent before enrollment.
The sponsor, Boehringer Ingelheim, designed and conducted the study in collaboration with the academic investigators. The sponsor monitored the study, collected data, and performed the statistical analysis. The academic investigators, participating institutions, and sponsor agreed to maintain confidentiality of the data. The first draft of the manuscript was prepared by the first author with support from a medical writer employed by the sponsor, and the first author made the decision to submit the manuscript for publication. All the authors had access to the data and assume responsibility for the integrity and completeness of the data and for the fidelity of this report to the study protocol.
Efficacy Assessments
The primary efficacy end point was a sustained virologic response (i.e., undetectable level of HCV RNA) 12 weeks after the completion of therapy. The secondary efficacy end points were the time to an undetectable level of HCV RNA, an undetectable level of HCV RNA at week 4 of treatment, and an undetectable level of HCV RNA 24 weeks after the completion of therapy.
Plasma HCV RNA levels were measured with the use of the quantitative COBAS TaqMan HCV High Pure System assay, version 2 (Roche), with a lower limit of quantification of 25 IU per milliliter and a lower limit of detection of 17 IU per milliliter. During treatment, HCV RNA levels were measured on days 1 and 4; at weeks 1, 2, 4, 6, and 8; and every 4 weeks thereafter. After the end of the treatment period, HCV RNA levels were measured at 4, 8, 12, and 24 weeks.
Safety Assessments
Biochemical and hematologic assessments were performed at each visit during the treatment period and 4 weeks after the last dose of the study drug was administered. Data on adverse events were obtained at each treatment visit and at the follow-up assessment. Physical examinations were performed at the screening visit, at the safety follow-up assessment, and as needed for the assessment and treatment of symptoms during treatment visits. An independent data and safety monitoring committee conducted regular planned reviews of the safety data.
Drug-Resistance Assessments
Plasma samples from all visits at which levels of HCV RNA were measured were stored for monitoring of drug resistance. HCV nonstructural protein 3 (NS3) and NS5B regions were sequenced in all patients at baseline and in patients who had virologic breakthrough during treatment or relapse of infection in order to identify viral variants associated with resistance to NS3/4A or NS5B inhibitors.
Statistical Analysis
The primary efficacy and safety analyses were based on the intention-to-treat population (all randomly assigned patients who received at least one dose of study medication). The proportion of patients with a sustained virologic response 12 weeks after the completion of therapy was calculated and compared among treatment groups with the use of a stratified Cochran-Mantel-Haenszel test to adjust for HCV and IL28B genotypes.16 Prespecified pairwise comparisons were made among the groups that received deleobuvir three times daily (to assess duration), between the BID28W and TID28W groups (to assess the deleobuvir dosage), and between the TID28W and TID28W-NR groups (to assess the effect of ribavirin). Post hoc pairwise comparisons of rates of a sustained virologic response 12 weeks after the completion of therapy were calculated within each treatment group for genotype 1a versus 1b (with adjustment for the IL28B genotype) and for IL28B CC versus non-CC (with adjustment for the HCV genotype 1 subtype). All P values reported for statistical comparisons are for descriptive purposes only; no significance threshold was prespecified, and no adjustments for multiple comparisons were performed. Ten post hoc subgroup comparisons were performed (the genotype 1 subtype and IL28B genotype within treatment groups). If there were no true differences between subgroups at a 0.05 significance level, the chance of at least one false positive result would be approximately 40%.
We also performed a multivariate logistic-regression analysis to evaluate the effects of covariates (age, sex, body-mass index, presence or absence of cirrhosis or diabetes, baseline alanine aminotransferase and γ-glutamyl transferase levels, viral subtype, IL28B genotype, and baseline HCV RNA level) on virologic response rates. This analysis was based on the per-protocol population, which excluded patients who prematurely discontinued the study therapy for reasons other than lack of efficacy (i.e., they had adverse events leading to discontinuation, were lost to follow-up, or withdrew consent).
Results
Patients
Of 469 patients who were screened, 362 underwent randomization and received at least one dose of the study drugs (Fig. S1 in the Supplementary Appendix, available at NEJM.org). Enrollment in the TID28W-NR group was discontinued on February 3, 2011, at the request of the Food and Drug Administration after other studies showed that virologic breakthrough was more common with interferon-free regimens that did not contain ribavirin than with those that did.13,17 Baseline demographic and clinical characteristics were similar in the five study groups.
Efficacy
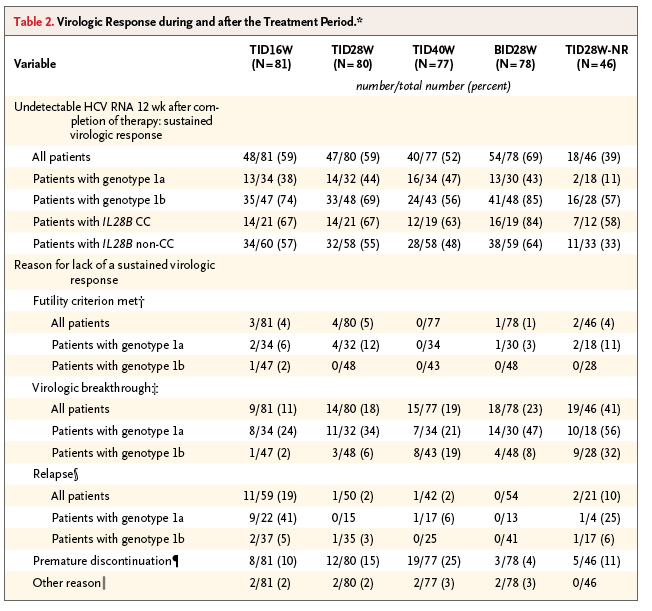
Virologic response rates are shown in Table 2 and in Table S1 in the Supplementary Appendix. There were no significant differences among rates of a sustained virologic response 12 weeks after the completion of therapy according to treatment duration (59% in the TID16W group vs. 59% in the TID28W group, P=0.86; 59% in the TID16W group vs. 52% in the TID40W group, P=0.37; and 59% in the TID28W group vs. 52% in the TID40W group, P=0.46) or deleobuvir dosage (69% in the BID28W group vs. 59% in the TID28W group, P=0.15). Rates of a sustained virologic response 12 weeks after the completion of therapy were higher among patients who received ribavirin than among those who received the same regimen without ribavirin (59% in the TID28W group vs. 39% in the TID28W-NR group, P=0.03).
Rates of a sustained virologic response 12 weeks after the completion of therapy were higher among patients with HCV genotype 1b infection than among those with HCV genotype 1a infection, after adjustment for IL28B genotype, in all groups except the TID40W group (BID28W, P<0.001; TID16W and TID28W-NR, P=0.001; TID28W, P=0.03; and TID40W, P=0.38). Response rates were also higher among patients with the IL28B CC genotype than among patients with non-CC genotypes in the BID28W and TID28W-NR groups (P=0.05 and P=0.02, respectively) after adjustment for viral subtype.
Genotype 1b, IL28B CC genotype, female sex, treatment regimens containing ribavirin, and normal baseline γ-glutamyl transferase levels were associated with a higher rate of sustained virologic response 12 weeks after the completion of therapy in the multivariate analysis (Table S2 in the Supplementary Appendix).
Fifty of the 75 patients who had virologic breakthrough during the study had HCV genotype 1a infection. Relapse occurred in 19%, 2%, 2%, 0%, and 10% of patients in the TID16W, TID28W, TID40W, BID28W, and TID28W-NR groups, respectively. Nine of 11 patients with relapsed infection in the TID16W group had HCV genotype 1a infection (Table 2).
A total of 97% of patients with virologic breakthrough (73 of 75 patients) had variants that emerged with mutants in both NS3 and NS5B, whereas the variants from patients who had relapsed infection were most commonly associated with single mutants in NS3 or NS5B (Table S3 in the Supplementary Appendix).
Safety
The most common adverse events were nausea, diarrhea, vomiting, jaundice, pruritus, rash, photosensitivity reaction, dry skin, asthenia, and fatigue (Table 3. Across all groups, 340 patients (94%) had adverse events and 34 (9%) had severe adverse events (defined as events that were incapacitating or led to an inability to work or perform usual activities). Between 56 and 73% of all the episodes of vomiting in the five treatment groups occurred during the first week of treatment. A similar pattern was observed for other gastrointestinal adverse events. Rashes were generally maculopapular and were typical of drug-related rashes. Photosensitivity reactions were commonly reported as exaggerated sunburns. Serious adverse events (defined as events that resulted in death, were life-threatening, resulted in persistent or clinically significant disability or incapacity, required or prolonged hospitalization, or were deemed to be serious by the investigator for any other reason, as detailed in the protocol) occurred in 7% of all patients (27 of 362). Of the patients with the most frequently reported events categorized as serious, 7 patients had skin disorders, 3 had infections, and 2 had anemia (Table S4 in the Supplementary Appendix).
The mean reductions in hemoglobin levels were 1.0 g per deciliter in the TID28W-NR group and approximately 2.5 g per deciliter in all other groups (Table 4. Anemia was reported as an adverse event in up to 13% of patients in the groups that received ribavirin and was not reported in any of the patients in the TID28W-NR group. Mean platelet levels increased from baseline during treatment in all groups (Table 4).
Discussion
The rates of a sustained virologic response 24 weeks after the completion of therapy among patients with previously untreated chronic HCV genotype 1 infection who received the current standard of care (pegylated interferon, ribavirin, and telaprevir or boceprevir) ranged from 68 to 75% in phase 3 trials.18-22 In this phase 2b study involving patients who had not previously received treatment for HCV genotype 1 infection, the combination of faldaprevir, deleobuvir, and ribavirin resulted in rates of a sustained virologic response of 52 to 69% 12 weeks after the completion of therapy.
Overall response rates were not affected by the treatment duration. However, the rate of relapse among patients with HCV genotype 1a infection who were treated for 16 weeks was higher than the rates among patients treated for 28 or 40 weeks with the same regimen (41% vs. 0% and 6%). Relapse rates among patients with genotype 1b infection were consistently low among all treatment groups (0 to 6%); these findings suggest that 16 weeks of treatment may be sufficient for this population.
The difference in response rates according to the deleobuvir dosage was not significant in this study (69% in the BID28W group vs. 59% in the TID28W group, P=0.15). However, the rate of premature discontinuation among patients who did not have a sustained virologic response 12 weeks after the completion of therapy was higher in the TID28W group than in the BID28W group (15% vs. 4%).
The low response rate observed in the group of patients who did not receive ribavirin was associated with high rates of virologic breakthrough and relapse. This finding is consistent with results from other studies of interferon-free regimens without ribavirin.13,17
The higher rate of a sustained virologic response 12 weeks after the completion of therapy among patients infected with HCV genotype 1b than among those infected with genotype 1a may be due to the fact that deleobuvir is less active against genotype 1a.23,24 In addition, there may be a lower barrier to the emergence of resistant variants for genotype 1a virus.25 Similar results were observed with other interferon-free regimens, indicating that genotype 1a may be more difficult to treat than genotype 1b.26,27
The observation that the IL28B polymorphism (rs12879860) affected the rate of a sustained virologic response 12 weeks after the completion of therapy suggests that innate immunity may still be important in interferon-free regimens.
However, the effect of the IL28B genotype is unclear in other interferon-free regimens.11,28
Adverse events were common; overall, 94% of patients had adverse events, and 9% had severe adverse events. Gastrointestinal and dermatologic adverse events were the most frequently reported events in this study. Patients received loading doses of faldaprevir and deleobuvir on the first day of the study to ensure sufficient plasma levels of both drugs during the first days of treatment. These loading doses may have contributed to the higher frequency of gastrointestinal events during the first week of treatment than later in the treatment period. Jaundice due to increased levels of bilirubin from baseline levels was observed in this study. Faldaprevir inhibits bilirubin metabolism primarily through inhibition of UGT1A1,29 which can lead to increased plasma levels of unconjugated bilirubin. In all cases of grade 3 or 4 hyperbilirubinemia, no concurrent increase in the alanine aminotransferase level was observed. The lower rate of jaundice in the TID28W-NR group (4%) than in the other groups can be explained by the absence of hemolysis in a ribavirin-free treatment.
Substantial reductions in red-cell, white-cell, and platelet counts are the most prohibitive side effects of interferon-based treatments for HCV infection. In the Individualized Dosing Efficacy vs. Flat Dosing to Assess Optimal Pegylated Interferon Therapy (IDEAL) study,30 30% of patients treated with pegylated interferon alfa-2a and the same dose of ribavirin used in this study had hemoglobin levels of less than 10 g per deciliter (i.e., anemia). In this study, 10% of patients in the groups that received ribavirin had hemoglobin levels that were less than 10 g per deciliter. The lower rate of anemia in this study might be due to the absence of the myelosuppressive effects of pegylated interferon; however, without a comparative study, this observation should be interpreted with caution. The low rate of anemia and the absence of a detrimental effect of the interferon-free regimens on white-cell or platelet counts may be an advantage of these regimens over interferon-based therapy.
This study has some limitations. First, the open-label design may have biased the comparative evaluation of the duration of treatment, since the patients who received longer regimens may have been more prone to early discontinuation of treatment than the patients who received shorter regimens. This may have contributed to the higher rate of discontinuation in the TID40W group.
Furthermore, the lack of an interferon-based control group limits the interpretation of the results in relation to the current standard-of-care treatment.
In conclusion, the interferon-free combination of faldaprevir and deleobuvir with ribavirin for the treatment of HCV genotype 1 infection was effective. The presence of ribavirin was found to be a necessary component of these regimens. The BID28W regimen had a more favorable efficacy and safety profile than did the other regimens examined in this study. The HCV genotype 1 subtype and host factors were found to be significantly associated with a sustained virologic response 12 weeks after the completion of therapy.
Supported by Boehringer Ingelheim.
|
|
| |
| |
|
|
|