| |
6th Intl Workshop on HIV Persistance during Therapy
|
| |
| |
Report Summary Report by David Margolis MD, UNC Chapel Hill and the Collaboratory of AIDS Researchers for Eradication (CARE)
The Sixth International Workshop on HIV Persistence during Therapy took place at the Marriott Marquis in Miami, rather than the sunny, sandy climes of St. Maarten, the site of this workshop every other year since 2003. Started a decade ago as a boutique meeting of interest to a few, the meeting was larger than ever before, and packed (perhaps overly so) with 67 oral presentations in two and a half days.
Bob Siliciano (Hopkins) opened the meeting with a plenary talk on the first evening outlining "Challenges In HIV Eradication Research." He outlined some of the many contributions that his group has made to the understanding of latent, persistent HIV infection of resting CD4+ T cells. He highlighted the recent work of Yan Chi Ho et al. in Cell, demonstrating the presence of proviral genomes that are not detected following a single round of in vitro activation in the gold-standard quantitative viral outgrowth assay (QVOA) that most accurately measures the size of the latent reservoir. Siliciano made the point that the demanding QVOA represents a definitive but perhaps minimal measurement of the presence of latent infection, as some "non-induced proviruses" can be recovered by PCR amplification and genetic reconstruction, or by additional cell activation and culture in the lab. Next he showed data from another recent publication showing that in a novel cell culture system, most current studied latency reversing agents contemplated for use in "shock and kill" approaches result in very little virus expression when compared to maximum T cell activation, suggesting that we have a long way to go before we will have potent, safe, and effective anti-latency therapy. He cited older evidence that even after successful induction of HIV gene expression, infected cells do not die from viral cytopathic effects and are not lysed by cytolytic T lymphocytes (CTL) from most patients on antiretroviral therapy. And finally he suggested that viruses with escape mutations in major CTL epitopes dominate the population of the latent reservoir. Most of the audience felt that it was a very pessimistic note on which to open the meeting, but perhaps as the cure field exerts a strong selective pressure for glass-half-full researchers, nobody packed up and went home. This optimism was borne out as for most of the rest of the meeting, various presentations showed that labs all over the world are hard at work at all of these challenges.
Mechanisms of HIV latency
Jon Karn (Case Western) opened the first session on the basic mechanisms of HIV latency by discussing the output of his group's effort perfomring shRNA library screens in Jurkat T-cells models of HIV latency to discover gene products that affected HIV latency. Many of the genes discovered were associated with chromatin silencing mechanisms. A novel target, the estrogen receptor ESR1, was discovered and both this receptor and its downstream signaling target, SRC3, are under study as potential targets of anti-latency therapy. Preliminary studies suggest that gossypol, an inhibitor of SRC3, can disrupt latency both alone and in synergy with the HDAC inhibitor vorinostat (SAHA or VOR).
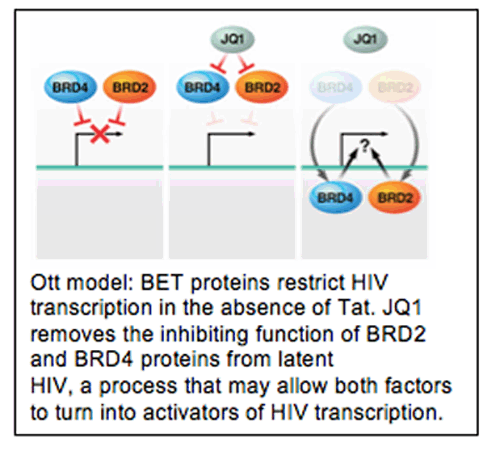
Melanie Ott from UCSF outlined a flurry of work from her lab, and several others, that suggested that the bromodomain family of transcriptional regulator proteins might be targeted to disrupt HIV latency. Like the histone deacetylase inhibitors, the bromodomain (BRD) family is large and complex, with multiple factors for multiple function. BRD proteins have the potential to inhibit some steps of HIV expression and activate others. Like HDAC inhibitors, numerous inhibitors of different specificities and selectivities exist. Many are potential clinical tools, and serve as scaffolds for the development of human drugs, such as the molecule JQ1.
Guido Poli (Institut San Raffaele) suggested that Class II histone deacetylases, not thought to have a direct role in HIV latency, might actually be a useful target to disrupt HIV latency as during stress or starvation, the Class II HDAC4 travels to the cell nucleus and affects the Class I HDACs known to be a key anti-latency target. Poli suggested the potentially synergistic use of class I and II HDAC inhibitors for reactivating latent proviral reservoirs. Carine van Lint (Bruxellles) outlined previous studies on the importance of the cellular cofactor CTIP2 for latency in a microglial cell model, a potential brain reservoir of virus. She suggested that CTIP2 has a double impact on HIV-1 latency, by both recruiting chromatin-modifying enzymes to enforce latency, and by inhibiting P-TEFb function, an activator required to escape latency. Stephan Emiliani (Paris), pointed to a complex of factors involved in viral integration, LEDGF/p75 associated with the cellular factors Spt6 and Iws1, as involved in the post-integration silencing of HIV the establishment and maintenance of HIV latency. And finally D. Alvarez from the Karn lab presented evidence that pharmacological inhibitors of the CoREST complex chromatin-modifying enzymes LSD1 and G9a/GLP could be of use in the treatment of HIV-infected microglial cells, but in this case to block activation and HIV expression, and potentially to prevent HIV-induced CNS toxicity.
Warner Greene (Gladstone) took the discussion in a different direction, with new data suggesting that a cell destruction pathway known as pyroptosis ("apoptosis by fire" or cell death with inflammation) is triggered by HIV infection, driving both CD4 T-cell death, and chronic inflammation that may drive cell proliferation and thereby allow latently infected cells to divide without being destroyed or expressing HIV. This very complex model that is sure to attract further study. Greene's lab studied human lymphoid aggregated cultures (HLACs) prepared using tonsil and spleen tissue from consenting HIV-infected volunteers not on ART. Productive HIV infection in activated CD4 T cells from tonsil and spleen promoted caspase-3-mediated apoptosis, but abortive infection of nonpermissive resting CD4 T cells (the majority of cells in this tissue) leads to by caspase-1-mediated pyroptosis, an intensely inflammatory form of programmed cell death. These events combine to create a vicious pathogenic cycle where dying CD4 T-cells release inflammatory signals that attract more cells to become abortively infected and die by pyroptosis causing more inflammation. Greene suggested that inhibitors such as VX-765, a small-molecule inhibitor of caspase-1 shown to be safe in humans, be tested in clinical studies.
Steve Deeks (UCSF) presented an overview highlighting the potential contributory role of persistent inflammation and immune dysfunction. The model suggests that a vicious cycle might exist in which HIV persistence causes inflammation that in turn contributes to HIV persistence. Remi Fromentin of the Chomont group at VGTI Florida showed some data consistent with this model, in which 8 different markers were measured on PBMCs from 48 virally suppressed subjects. Three of these 8 markers, cellular receptors that transmit negative regulatory signals, PD- 1, LAG-3 and TIGIT, were associated with incomplete CD4 T-cell restoration and HIV persistence as measured by HIV DNA. Given the variable association of HIV DNA with true persistence, this is an interesting but not definitive observation. Mailliard of the Rinaldo group at U Pitt proposed that certain cytotoxic T lymphocyte (CTL) responses to HIV became dysfunctional, in that they provided an inflammatory response to did not kill or clear HIV. Such cells might paradoxically enhance cell-to-cell HIV infection and thereby allow persistence of infection.
Assays to Measure HIV Persistence
Janet Siliciano opened this discussion with by detailing a recent collaborative study comparing 11 different approaches for quantitating persistent HIV-1. Assays were compared to the gold-standard quantitative viral outgrowth assay (QVOA) that measures the frequency of the recovery of replication-competent HIV from circulating resting CD4+ T cells. Various PCR-based assays of either PBMCs or resting CD4+ T cells for total HIV- 1 DNA, or specifically for integrated HIV, circularized proviral episomes, or HIV DNA in rectal cells gave infected cell frequencies at least 2 logs higher than the viral outgrowth assay, even in subjects who started ART during acute/early infection. These DNA measures were not even useful on a per-patient basis for anything but a general categorization (ie "a whole lot" of latent infection or "not so much") as the ratio of infected cell frequencies determined by viral outgrowth to any of the PCR-based assays varied dramatically between patients. The dramatic differences in infected cell frequencies and the lack of a precise correlation between culture and PCR-based assays means that even if a patient was actually cured of HIV infection, DNA might still be detected, or that if a patient had undergone a procedure that depleted 50% of the latent reservoir, it might not register on a DNA assay. Certainly such assays could still be useful as they are so simple, and might give a signal of some very substantial effect (which is what is ultimately needed), but they emphasize the fact known to retrovirologists for decades that most of the detectable HIV DNA is non-functional. To be provocative but to make a long-ignored point, it is formally incorrect and perhaps even intellectually dishonest at this point to claim that we are "measuring the HIV reservoir" when any PCR measure of HIV DNA is done. The viral rebound of the "Boston patients" (more on that later) who showed huge declines of HIV DNA prove that point yet again.
This is not to say that the QVOA is perfect. There is no free lunch in this business, it seems. Janet Siliciano again discussed the work of Ho et al., reviewing the data that showed that in about half of the handful of patients studied thus far, the QVOA assay appears to represent the "true reservoir" relatively well, but in the other half of patients a large excess of genomes can be detected (5-fold to 60-fold) that express HIV RNAs without major genetic defects and that appear to be able to replicate in culture systems. The glass-half-empty view is that "the barrier to cure-may be up to 60-fold greater than previously estimated." The glass half full view is that it is not so in a lot of patients, and in the others the total number of cells is still less than that of a small, removable tumor. The challenge of course, is getting ALL of the tumor out in a reasonable period of time, in a safe and affordable way. Siliciano did mention that in preliminary studies, about 25% of the viruses that are not recovered from cells after a single round of stimulation in the lab, do emerge in culture after a second round of stimulation. So part of the answer may be serial purging of the reservoir.
Three presentations then followed describing various uses of the emerging "digital droplet" PCR technique to quantitate HIV RNA and DNA forms. Strain (UCSD), Hu (Brigham and Women's), and De Spiegelaere (Ghent) discussed this new technique that is based on performing ddPCR in a solution portioned by microdroplets. The instruments generate thousands of picoliter-sized droplets that contain PCR reaction mixtures and (on average due to dilution of the sample) no more than one target DNA or RNA molecule per droplet.
This allows ddPCR to assay millions of cells and to be very more precise at low copy number. Strain showed data measuring unspliced HIV Gag (a late mRNA), multiply spliced Tat-Rev RNA (an early mRNA), and full-length RNAs encoding the poly A tail. Various technical issues are still to be ironed out, including noise signal at the lower limits of the assay (more troublesome when measuring HIV RNA than HIV DNA), variability of RNA extraction depending on the kit used, and viral target sequence variation issues, among others. But the ddPCR technology appears to be a promising new tool for the quantification of HIV-1 cell-associated DNA and RNA.
Sarah Palmer, late of the Karolinska but now at the Westmead Millennium Institute and University of Sydney, studied the genetic makeup of HIV DNA in cells over time. Whereas the frequency of HIV DNA does not give a very good measure of the frequency of true replication-competent infection, the changes of the population of HIV DNA sequences over time does provide definitive proof that HIV was growing in the cell studied in the past, as this is the only way this RNA virus leaves a DNA footprint. Using single-genome and single-proviral sequencing techniques, Palmer's lab isolated intracellular HIV-1 genomes derived from defined subsets of T cells (naïve, central-, transitional-, and effector-memory) from peripheral blood, marrow, GALT, and lymph node tissue. Samples were collected at 2 time points (separated by 6 months) from 8 subjects on suppressive therapy (4-12 years): 5 who initiated therapy during acute infection and 3 who initiated therapy during chronic infection. Maximum likelihood phylogenetic trees were constructed using the general time reversible model. Looking at the frequency of specific DNA sequences over time, and the changes seen in DNA sequences over time, the expansion of some HIV genetic populations and the contraction of others with little evidence of viral evolution --- that is more cells contained specific sequence population "X" and specific sequence population "Y" disappeared or diminished in proportion. As an example in one patient a clonal sequence species containing a large 380 base pair deletion was dominant, and increased from 71% to 92% of the sequences found over 6 months in peripheral blood effector memory T cells. The results were consistent with a model in which the pool of HIV-infected resting memory CD4+ T cells typically does not change dramatically over 6 months in different tissue compartments. The increase of clonal HIV-1 sequences, especially a large deletion mutant, indicates an expansion of cells with dead, defective proviral DNA rather than active viral replication.
Maldarelli (NIH Frederick) described the case of a single patient with an oral carcinoma who developed persistent low-level viremia 200-300 c/ml after 11 years of suppressive ART. Single-genome sequencing (SGS) revealed both WT and multidrug-resistant HIV. The WT population consisted largely of multiple identical sequences; the drug-resistant population comprised diverse variants encoding K103N and M184V. Switch of ART to Truvada/RAL produced a 10-fold reduction of the drug-resistant variants, leaving almost only the WT variants. The emergence of clonal, WT viremia on cART and its insensitivity to cART implies that the source of viremia was an expanded clone of HIV-infected cells perhaps including increased HIV production from that clone.
The same group investigated the distribution of HIV infected cells within gut mucosa by analyzing endoscopy-derived material. Representative biopsy samples from 7 patients were obtained throughout the colon in all patients and from ileum in 4/7 patients. Single genome sequencing showed that HIV proviruses are extensively and uniformly distributed throughout the GALT without clear evidence of sampling variation. Clonally expanded populations present in gut mucosa were not anatomically restricted, and were not divergent from pre-therapy HIV. Maldarelli and colleagues concluded that detection of the same identical sequences in gut derived DNA and RNA, and in plasma suggests that GALT could be one source of persistent viremia on ART. But while this conclusion is potentially true, it could equally be true that persistently infected cells, perhaps originating from a proliferating founder population, could traffic through the GALT where they encounter environmental activation signals and produce RNA but not necessarily spreading infection/replicating virus in the face of ART.
Laboratory and animal models of persistent HIV infection
Several presentations focused on laboratory and animal models of latent HIV infection, a critical need as fully validated models are still lacking. Vicente Planelles (Utah) opened with a presentation from the CARE collaboratory (nb. The author is a member and a co-author for this presentation) that compared the reactivation of latent HIV in virtually all the primary cell models of latent HIV infection that have ever been described in the world's literature. The group sought an answer to a burning question --- what is the best cell culture model system for replication latent HIV infection in resting CD4+ T cells in vivo? Although major advances have been facilitated by the use of latently infected T cell lines, these are usually criticized as artificial, and the newer primary cell models in which primary cells are infected and then forced in to the latent state are felt to be preferred. However, notable differences exist among cell model systems. Furthermore, screening efforts in specific cell models have identified drug candidates for "anti-latency" therapy, which often fail to reactivate HIV uniformly across models. Therefore the group compared the responses of five primary T cell and four J-Lat cell line models to that of the standard viral outgrowth assay using patient-derived infected cells (QVOA) across a panel of thirteen stimuli that are known to reactivate HIV by defined mechanisms of action. Maddeningly no single cell model system was able to perfectly capture the ex vivo response characteristics of latently infected T cells from patients. Specific model systems appear biased in favor or against certain signaling pathways. The data is very complex, and will be published soon in PLoS Pathogens. It should allow investigators to select the best model, or group of models, for future studies and allow them to choose models responsive to certain types of signals, if desired.
Janice Clements (Hopkins) reported progress in the study of the SIV macaque model, reporting the use of a quantitative viral outgrowth assays (QVOA) for SIV, quantitating SIV within monocytes, tissue macrophages and microglia. Viremia was been suppressed to low levels (< 10 copies per ml plasma) with two different cART regimens and we have used this model to develop a QVOA assay for monocytes and tissue macrophages. Within the CD11B+ cell population the authors found 5.2 cells/million were vRNA positive, despite ART suppression. CD11b is expressed on the surface of many leukocytes including monocytes, neutrophils, natural killer cells, granulocytes and macrophages, as well as on 8% of spleen cells and 44% of bone marrow cells, but not T cells.
Koen von Rompay (UC Davis) then gave an update on effort in the Luciw laboratory to model the effect of the HDAC inhibitor SAHA (vorinostat) on viral reservoirs in ART-suppressed nonhuman primates infected with the RT-SHIV virus construct. This study could validate the primate model by reproducing results seen in human studies of SAHA. Juvenile macaques are started 6 weeks after intravenous RT-SHIV inoculation on an ARV regimen (tenofovir, emtricitabine, efavirenz once daily) known to rapidly reduce plasma viremia. Once plasma virus levels will have reached very low or undetectable levels, pulsatile treatment with subcutaneous injections of SAHA was begun. The biological effect of the drug, histone acetylation, has been observed in peripheral blood cells. Measurements of the effect of this treatment on HIV reservoirs and HIV RNA expression in both blood and tissue samples is ongoing, with results expected in early 2014.
J Victor Garcia (UNC) then discussed the other animal model of persistent HIV infection, presenting a systemic examination of the latent and residual active HIV reservoirs in bone marrow-liver-thymus (BLT) humanized mice undergoing ART. He and his coworkers found that, as in humans, the latent HIV reservoir is broadly disseminated during ART, and that human tissues examined throughout the mouse exhibited low level vRNA production -- indicating that the residual active HIV reservoir is also systemic in nature. The BLT appears capable of providing a quantitative framework for the evaluation of the in vivo efficacy of HIV eradication interventions designed to deplete HIV. Garcia then presented two new modifications of the BLT model, the TOM (T cell only) and the MOM (macrophage only) mouse. HIV replication, ART suppression, viral rebound after ART interruption, and resting cell latent infection were all seen in the TOM. Studies of the MOM are in earlier stages, but low-level HIV replication after infection has been observed. These exciting new tools may allow the dissection of the role of each of these cell types in HIV persistence in vivo.
Following this, Hans Peter Kiem from the defeat HIV collaboratory reported on studies in two monkeys (compared to 2 controls) in whom ex vivo transduction of their stem cells with the mC46 HIV fusion inhibitor construct appeared to produce gene-protected stem cells. Monkeys given these cells then had CD4 preservation after SIV infection.
Jeff Lifson (NCI Frederick) furthered the discussion of NHP model systems, suggesting that major gaps in our understanding of viral reservoir establishment, maintenance, phenotype and tissue compartmentalization that are particularly difficult to study in humans can be readily studied in NHP. He illustrated various techniques for visualizing HIV RNA in cells and tissues. He highlighted 3 techniques to detect SIV RNA: 1) radiolabelled in-situ hybridization (ISH), which is slow (5-14 days), and subject to high tissue background, 2) positive-strand viral RNA ISH with chromogenic detection, quicker (3 days) but with poorer resolution, and 3) RNA SCOPE ISH, which is quick (8 hrs), of low background, and potentially may be able to distinguish single virions.
Pharmacology of HIV Persistence
Two years ago at the last St. Martin meeting, Courtney Fletcher (U Nebraska) presented high-profile data suggesting widely inadequate ART drug concentrations in the tissues. His presentation of data from a continuation of the same study 2 years later seemed more muted. He hypothesized that antiretroviral drug concentrations in lymphoid tissue might be insufficient to fully suppress replication in these sites. 12 patients were followed since ART initiation, with multiple samplings of lymph node, ileum and rectum, and peripheral blood, to determine intracellular concentrations of the ARVs in these tissues and to measure levels of persistent HIV. He restated some findings from 2 years ago, that concentrations of some frequently used drugs (tenofovir, FTC, efavirenz, atazanavir) are much lower in lymph nodes than in peripheral blood, but darunavir and raltegravir levels were adequate. However, TDF and FTC levels were higher in rectal and ileal tissue than plasma. All drug levels were adequate in the rectum. These data still require a fuller presentation and publication, with internal standards and controls to insure that drug levels did not drop during sample processing. Fletcher did find that on a per-patient basis, higher drug concentrations correlated with more rapid decline in HIV RNA within the follicular dendritic cell network.
Angela Kashuba (UNC) discussed similar issues, and agreed that limited data exist to evaluate inter-species similarities or differences in extracellular or intracellular drug distribution in tissue sites. She had similar comments on ARV concentrations in animal models. She outlined new methods to evaluate the relationship between pharmacology and HIV persistence, and noted the challenge of tissue homogenization LC-mass spec) without losing sample.
Jay Grobler from Merck presented arcane-sounding work entitled "Inhibitory Slopes Show Minimal Variation Within and Across Mechanistic Classes of HIV-1 Antiretroviral Agents and Are Not Likely to Contribute to Differential Effectiveness of Combination Therapy and Viral Persistence." The analysis stems from work in the Siliciano laboratory, which suggested that the slope of the ART concentration inhibition curve (Hill coefficient) is an important factor in selecting the best ART combinations. Grobler showed that this was true when assays of drug effect were performed in a specific cell line, 293T, as had been done in the Siliciano work, but not when assays were run in peripheral blood mononuclear cells (PBMCs) and lymphoid-derived cell lines. He concluded that consistent with clinical experience, the slopes of dose response curves are not likely to be a significant factor in determining clinical efficacy or contribute to the effectiveness of drug specific combinations.
Drug Development and Testing Curative Strategies
Daria Hazuda presented an overview of the Merck program. She described the most advanced of these approaches, the use of histone deacetylase inhibitors (HDACi), and recent studies suggesting the actions of these inhibitors might be complex and unique. HDACis with similar inhibitory potencies can have widely different enzyme binding kinetics, and the potency of HDAC inhibition might not be the most important measure of a compounds utility against HIV latency. She outlined a high-throughput screen that had discovered many new potential anti-latency compounds, many of which appeared to increase in potency when used with the HDACi SAHA (vorinostat). She introduced the discovery of the activity of farnesyl-transferase inhibitors (FTIs) as anti-latency reagents. Richard Barnard from Merck went into this work in detail in a subsequent talk, outlining the characteristics of these compounds, previously developed as (unsuccessful) anti-cancer drugs. FTIs result in modest induction of the expression of HIV in model systems and in cells from patients, but substantially increase the magnitude of induction in combination with SAHA, as well as more selective HDACi (against specific HDAC isoforms), and with other reagents such as prostratin. FTIs of two different mechanistic classes are active, and activity correlates with FTI potency, suggesting that mode of induction is directly related to inhibition of FT. Data suggested the potential for increased efficacy in combination and providing proof of concept for identifying synergistic combinations using this novel screening paradigm.
Suzanna Valente of the Scripps Research Institute in Florida discussed her recently published findings on a potent inhibitor of the viral activator Tat, didehydro-Cortistatin A (dCA), an analogue of a natural steroidal alkaloid from a marine sponge. Oppositely of most current approaches, this strategy would hope to establish a state of "super latency" of HIV, preventing viral reactivation from latently infected cells if ART was stopped. This has been achieved in cell model systems, and dCA inhibits HIV reactivation upon homeostatic and antigenic stimulation of CD4+T cells isolated from virally suppressed patients undergoing ART. The next step would be proof-of-concept studies in an animal model or in the clinic.
Stephen Mason outlined the BMS program, which included screening efforts to identify novel anti-latency compounds, but also emphasized immunomodulatory therapies designed to augment the anti-HIV immune response. As proof of concept, nivolumab (anti-PD-1; BMS-936558) has been tested in the context of chronic HCV in human subjects with modest effect. Alternatively, an alternate ligand of the same pathway, BMS-936559 (anti-PD-L1) has been administered to SIV-infected rhesus macaques that were suppressed on a combination of antiretrovirals (ARVs), and testing in man through the ACTG is in the planning stages. The effect of anti-PD-L1 immunotherapy in ARV-suppressed rhesus macaques was reported by James Whitney (Beth Israel Deaconess Medical Center). 13 MHC-defined rhesus macaques were confirmed SIVmac251 positive and ARV treatment was initiated, using a 4 drug ARV regimen, for a minimum of 6 months prior to the administration of BMS-936559. All 13 animals received either BMS-936559 (N=8) or isotype control antibody (N=5). Five doses of 10 mg/kg were given over 2 weeks, and effects measured at days 0-14. The repeated dosing of BMS-936559 was also well tolerated in all animals with no noted untoward effects. Receptor occupancy of BMS-936559 was favorable. ART was then continued for 6 weeks, and then an analytic treatment interruption was undertaken. In the BMS-936559-treated animals, viral rebound was lower than setpoint viremia in 7 of 8 animals, and 1.5 logs lower in 4 of 8 animals. Rebound was lower than setpoint in only 1 of 5 control animals.
Romas Geleziunas outlined the Gilead program, a comprehensive effort testing the HDACi rhomidepsin (ACTG study nearing initiation), other agents such as PKC agonists, and including a high-throughput screening effort.
Immunotherapeutics mentioned included therapeutic vaccines, and monoclonal antibody therapeutics designed to clear infected cells. Recent studies have identified a TLR7 agonist as an inducer of CD8 and NK cell response, and a combination approach using rhomidepsin and TLR7 in the rhesus model is planned.
Shytaj reported the findings published by the Savarino group (Rome) of a functional cure-like condition in chronically SIVmac251 infected macaques. A combination of ART, auranofin and buthionine sulfoximine (BSO) induced spontaneous post-therapy control of viral load in chronically SIVmac251-infected macaques [Shytaj et al., Retrovirology. 2013]. This control was associated with increased and broad anti-Gag immune responses and was dependent on the presence of CD8+ cells.
Lucio Gama and Janice Clements (Hopkins) described the use of Ingenol-B (ingenol-3-hexanoate), a potent PKC activator. A hexanoate derivative of Ingenol (Ing-B), a phorbol ester isolated from the Brazilian shrub Euphorbia tirucalli, Ing-B was given to SIVmac251-infected macaques, 0.4 mg/kg/day for 30 days. In the absence of ART Ing-B increases viremia. With PMPA, FTC, and RAL, ingenol increased plasma VL, but then after ART interruption, VL dropped to undetectable in some animals. A deeper tissue analysis of the effects of Ing-B is awaited
Keith Jerome of the defeat HIV collaboratory discussed the design and delivery of homing endonucleases designed to inactivate HIV provirus. Using a combination of designer HEs that target multiple essential HIV genes for disruption and subsequent inactivation we aim to inhibit the process of HIV inactivation from latently infected reservoir cells. We have tested this hypothesis in latently infected primary central memory T cells, by delivery of designer enzymes using scAAV vectors that can infect more than 90% of primary cultured Tcm.
Several groups are beginning to examine therapeutic vaccination as part of an eradication strategy, and the potential interaction of the host effects of anti-latency therapies on the immune response. Jones (Ragon Inst) used a model of latency resulting from the infection of primary CD4 T cells in culture, and examined the effect of CTL clones on these target cells. HIV-specific CTL clones were co-cultured with these targets in the presence or absence of latency-reversing agents. Interferon-gamma was quantified as a measure of CTL recognition. Jones found that while HDACi and common gamma-chain cytokines both reversed HIV latency, HDACis suppressed CTL function and thus performed poorly in integrated "flush and kill" assays. In contrast, IL- 15 superagonist (IL-15SA) both reversed HIV latency and enhanced CTL function. Treating patient CD4+ T-cells with a romidepsin pulse/wash followed by co-culture with HIV-specific CTL and IL-15SA resulted in a 5-fold reduction in levels of provirus and in the elimination of infectious virus as measured by viral outgrowth assays.
Julia Sung (UNC) from the author's laboratory presented the use of ex-vivo expanded CTLs as utilized for viral infections in oncology to clear latent HIV infection. As compared to unexpanded CD8s, expanded CTLs reduced p24 production from autologous targets superinfected with the lab virus JR-CSF (median %p24 produced with expanded CTLs=2.5%, vs 29.2% with unexpanded CD8s, p<.05) or autologous virus obtained from the patient's own latent reservoir (median 8% vs. 20.6%, p<.05). We feel that ex-vivo expanded CTLs could prove useful in combination with latency reactivating agents.
Human Studies
As you may have read in the paper, Tim Henrich of the Brigham and Women's Hospital, announced the unhappy news that both of the so-called "Boston patients" had rebounded with symptoms of primary infection. These patients had had marrow transplants for cancer, but had not received HIV-resistant cells. At 4.3 years after transplantation, one patient was completely transplanted, with only donor cells detected, and no HIV DNA in two pools of 25 million PBMCs that were tested, a reduction of HIV DNA of at least 1500-fold compared to the level of DNA prior to transplant. At 2.6 years after transplantation the second patient was completely transplanted, with only donor cells detected, and no HIV DNA in one pool of 50 million PBMCs that were tested, and no HIV recovered after the culture of 150 million PBMCs. Both patients had no detectable HIV-specific cellular immune function by ELISpot IFN-gamma screenings of total PBMCs involving comprehensive HLA-specific peptide panels. But they did have significant chronic graft-vs-host disease (GVHD), that is the patients engrafted immune systems were exhibiting a chronic active immune response against the patient's native cells. After the ART interruption 1 patient rebounded within 12 weeks (and just after the patients' cases were described at the summer IAS meeting) and the other 224 days after interruption. Of note, an assay for HIV DNA on day 196 was negative, but viremia recurred 28 days later. It is very unfortunate that, as far as we know at this point, a comprehensive analysis of a sufficient number of cells was not done to stringently analyze viral persistence prior to rebound.
Katherine Luzuriaga (UMass) reviewed the well-known case of the Mississippi baby, and the IMPAACT network's plans to attempt to replicate the case by finding and treating high-risk children within 48 hours of birth.
David Margolis (UNC, the author of this summary) presented an update from the ongoing studies of SAHA or vorinostat (VOR). The potent Class I HDACi VOR upregulates HIV RNA expression within the resting CD4+ T cells of ART-treated, aviremic HIV+ patients in vivo. But the ability of VOR to repeatedly disrupt latency is unproven, the optimal dosing schema is unknown, and the effect of VOR on host mechanisms that might clear infected cells is uncertain. In a Phase I-II single-center study, HIV+ participants maintained suppressive ART, and resting CD4+ T cells were obtained via leukapheresis. If an increase in resting CD4+ T cell-associated HIV RNA (RC-RNA) was measured following a single VOR 400 mg dose, patients received VOR 400 mg daily M-W for 4 weekly cycles, followed after a 4 week rest period by another 4 weekly cycles. Sparse VOR PK, biomarker measurements of histone acetylation within PBMCs, HIV RNA single-copy assays, RC-RNA, total cellular HIV DNA, and quantitative viral outgrowth assays (QVOA) from resting CD4+ T cells were obtained. In 5 patients VOR was well tolerated with no adverse events greater than Grade I; mild declines in platelet counts < Grade I were seen commonly. VOR exposures were within expected parameters. However, when measured after dose 11 (2nd dose of cycle 4) and dose 22 (2nd dose of cycle 8) cellular histone acetylation was little increased from baseline levels, and measures of RC-RNA only modestly increased in some patients. QVOA and other assays were also generally stable. We believe that complex feedback host mechanism blunt the response to repeated VOR doses if the dosing interval is too frequent.
Sharon Lewin (Melbourne) presented follow-up data from her study of daily VOR. She evaluated differential gene expression in blood from 20 HIV-infected patients on ART who received vorinostat (400mg/day) for 14 days. Gene expression was analyzed at baseline and two time points on vorinostat (d1 and d14) and post-cessation of vorinostat (d84). We found that the effect of vorinostat on chromatin largely occurred within the 1st day after the first dose of drug and that after 14 days of continuous dosing, there were compensatory mechanisms associated with transcriptional repression and cell survival. These results demonstrate significant effects of vorinostat on viral proteins and host genes that may have a significant impact on the potency of activation of latent virus.
Thomas Rasmussen also presented further data on the Aarhus Hospital's ongoing panobinostat studies. In a phase I/II clinical trial, 16 HIV-infected adults on suppressive ART received treatment with oral panobinostat 20 mg 3 times per week MWF every other week over the course of 8 weeks. 16 grade I adverse events were noted, 10 of 15 thought to be drug related, and mostly fatigue or GI upset. Although CD4 cells were stable, there was a mild drop in neutrophils and platelets, which reversed and did not achieve a gradable toxicity. An increase of histone acetylation was seen post-dosing but never seemed to return all the way to baseline. HIV cell-associated RNA increased variably, with a clear 2-fold increase on dose 1, but a variable increase of 1.5-2.5 fold after that.
Zeidan reported the results of the Sangamo trials of the zinc finger nuclease SB-728-T that edits the human CCR5 gene and confers HIV resistance to cells (as I believe had been reported at ICAAC).
In seven SB-728-T treated CCR5 Δ32 HIV subjects, VL decreased by >1 log from peak in three subjects after ART interruption. Two subjects achieved unmeasurable VL, in one subject from 11 through 19 weeks of TI and ongoing. VL reduction from peak correlated with the level of circulating bi-allelically CCR5 modified cells during TI (r=-0.81, p=0.015). As high levels of CCR5 modification, along with poly-functional CD8 anti-GAG responses, and low HIV-DNA levels in PBMCs appear to play an important role in functional control of HIV with SB-728-T treatment, the group seeks to pursue cytoxan chemotherapy pre-conditioning to "make room" in the marrow and enhance SB-728-T engraftment.
Macatangay (U Pitt) reported the results of a dendritic cell-based HIV therapeutic vaccination study. Autologous dendritic cell (DC) vaccine pulsed with autologous, inactivated HIV-1-infected apoptotic cells were given to 10 ART-treated subjects. After at least 8 weeks of virologic suppression, 6/9 subjects had residual viremia detected by a new single-copy assay (iSCA) ranging from 2.0-49.5 cps/mL. In 6/10 subjects, increasing levels of residual viremia were observed after vaccination despite continuous ART. Increased residual viremia was measured by iSCA in 40% of subjects despite continuous ART. This increase was not associated with increased T cell activation. iSCA is a more sensitive tool for detecting rebound viremia than the FDA-cleared Roche Amplicor assay v1.5. Therapeutic vaccination may increase HIV-1 replication or expression from latent reservoirs.
Finally, some evidence suggests the Wnt cellular signaling pathway plays a role in maintaining latency. Lithium, an inhibitor of Wnt signaling pathway, might synergize with HDAC inhibitors in inducing the reactivation of the latent HIV-1 LTR. Puertas (IrsiCaixa, Badalona) and colleagues nested an examination in to an ongoing clinical study designed to assess the effect of lithium on HIV-associated neurocognitive impairment, and explored lithium's potential effect on HIV-1 reactivation and viral reservoirs. Nine ART-suppressed subjects received treatment with lithium carbonate, beginning a 2-daily 400 mg dose, and changing further adjusting the dose according to drug levels in serum. Changes in total cell-associated HIV-1 DNA and RNA in circulating primary CD4+ T cells were estimated by droplet digital PCR at weeks 0, 2, 4 and 12 during treatment with lithium. The frequency of latently infected cells was also quantified by the viral outgrowth assay (IUPM) at weeks 0 and 12. The therapeutic administration of lithium in ART-suppressed subjects did not show any sign of viral reactivation or had a significant effect on the size of the HIV-1 reservoir. However, the Wnt pathway is complex, and the effect of lithium on the pathway may be bimodal. Alternative inhibitors or inducers that affect Wnt signaling might have a larger impact.
Overall, the workshop was exciting and packed with information. The HIV Cure research field is moving ahead, although the road is still a long one. Surely there will be more news at CROI this spring, and IAS this summer.
|
|
| |
| |
|
|
|