| |
Simeprevir plus sofosbuvir, with or without ribavirin, to treat chronic infection with hepatitis C virus genotype 1 in non-responders to pegylated interferon and ribavirin and treatment-naive patients: the COSMOS randomised study
|
| |
| |
Download the PDF here
The Lancet, Early Online Publication, 28 July 2014
"Patients were separated into two sequentially recruited cohorts. Those eligible for cohort 1 were required to be previous non responders to peginterferon and ribavirin and to have no to moderate liver fibrosis (METAVIR score F0-F2), and those eligible for cohort 2 were required to be previous non-responders to peginterferon and ribavirin or treatment naive and have severe liver fibrosis (METAVIR score F3-F4).......168 patients were enrolled and randomised, and 167 started treatment (n=80 in cohort 1 and n=87 in cohort 2). SVR12 was achieved in 154 (92%) patients (n=72 [90%, 95% CI 81-96] in cohort 1 and n=82 [94%, 87-98] in cohort 2)."
Eric Lawitz, Mark S Sulkowski, Reem Ghalib, Maribel Rodriguez-Torres, Zobair M Younossi, Ana Corregidor, Edwin DeJesus, Brian Pearlman, Mordechai Rabinovitz, Norman Gitlin, Joseph K Lim, Paul J Pockros, John D Scott, Bart Fevery, Tom Lambrecht, Sivi Ouwerkerk-Mahadevan, Katleen Callewaert, William T Symonds, Gaston Picchio, Karen L Lindsay, Maria Beumont, Ira M Jacobson
Summary
Background
Interferon-free regimens are needed to treat hepatitis C virus (HCV) infections. We investigated the efficacy of combined simeprevir and sofosbuvir.
Methods
We enrolled patients with chronic HCV genotype 1 infections who had previously not responded to pegylated interferon (peginterferon) and ribavirin or were treatment naive. Patients were randomly assigned in a 2:2:1:1 ratio to receive 150 mg simeprevir and 400 mg sofosbuvir daily for 24 weeks with (group 1) or without (group 2) ribavirin or for 12 weeks with (group 3) or without (group 4) ribavirin, in two cohorts: previous non-responders with METAVIR scores F0-F2 (cohort 1) and previous non-responders and treatment-naive patients with METAVIR scores F3-F4 (cohort 2). The primary endpoint was sustained virological response 12 weeks after stopping treatment (SVR12). Analysis was done by intention to treat. Safety data from cohorts 1 and 2 were pooled for analysis. This study is registered with ClinicalTrials.gov, number NCT01466790.
Findings
168 patients were enrolled and randomised, and 167 started treatment (n=80 in cohort 1 and n=87 in cohort 2). SVR12 was achieved in 154 (92%) patients (n=72 [90%, 95% CI 81-96] in cohort 1 and n=82 [94%, 87-98] in cohort 2). The most common adverse events in the pooled groups were fatigue (n=52 [31%]), headache (n=33 [20%]), and nausea (n=26 [16%]). Grade 4 adverse events were seen in one (2%) of 54 patients in each of groups 1 and 3 and in three (10%) of 31 patients in group 2, whereas grade 3-4 events were reported in less than 5% of all patients, except increased blood amylase concentration. Serious adverse events were seen in four (2%) patients, all in groups 1 and 2. Four (2%) patients discontinued all study treatment because of adverse events, three before week 12.
Interpretation
Combined simeprevir and sofosbuvir was efficacious and well tolerated.
Figure 2: Patients who achieved sustained virological response 12 weeks after the planned end of treatment in the intention-to-treat population, by treatment group (A) Cohorts 1 and 2. (B) HCV subtype and presence of the HCV Gln80Lys polymorphism at baseline in cohort 1. � HCV subtype and presence of the HCV Gln80Lys polymorphism at baseline in cohort 2. (D) HCV subtype and presence of the HCV Gln80Lys polymorphism at baseline pooled for cohorts 1 and 2. GT=genotype.
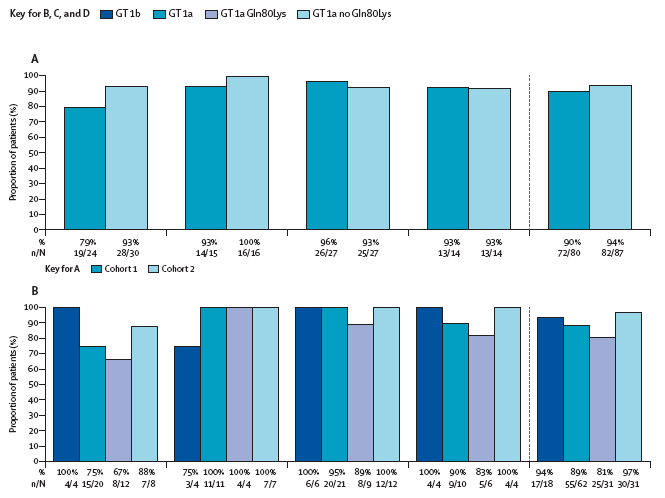
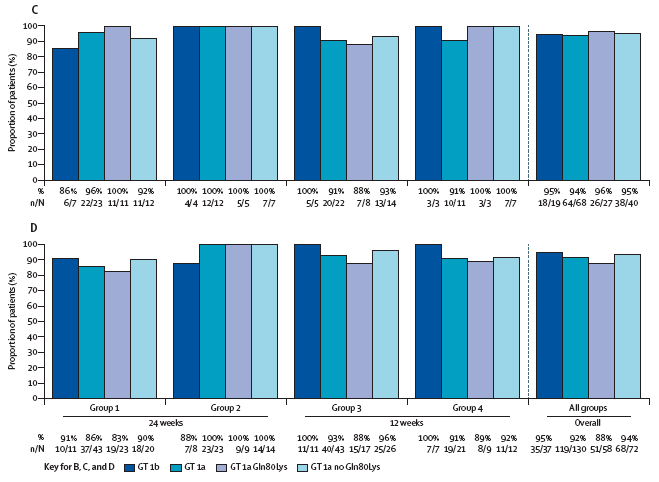
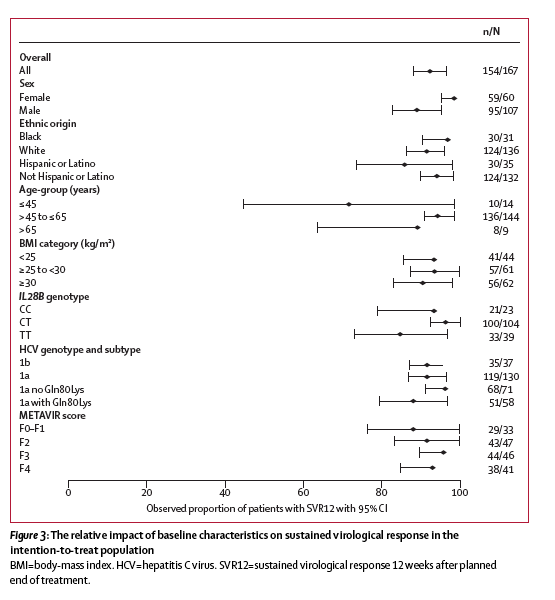
Figure 3: The relative impact of baseline characteristics on sustained virological response in the intention-to-treat population
BMI=body-mass index. HCV=hepatitis C virus. SVR12=sustained virological response 12 weeks after planned end of treatment.
Introduction
Chronic infection with hepatitis C virus (HCV) is a worldwide health problem that can lead to cirrhosis, decompensated liver disease, and liver cancer. 130-150 million people are chronically infected worldwide and 350 000-500 000 HCV-related deaths are reported annually.1 Treatment for HCV genotype 1 has evolved from pegylated interferon (peginterferon) and ribavirin2 to include direct-acting antiviral agents.
Simeprevir is a once-daily HCV NS3/4A protease inhibitor that is approved for use in combination with peginterferon or ribavirin to treat chronic infection with HCV genotype 1. In phase 3 studies done in patients with cirrhosis, sustained virological response (SVR) rates of 80-81% were reported in treatment-naive patients,3, 4 79% in relapsed patients,5 and 54% in partial or non-responders to previous peginterferon or ribavirin therapy.6 Sofosbuvir is a once-daily HCV nucleotide-analogue NS5B polymerase inhibitor that is approved for the treatment of chronic infections with HCV genotypes 1-4. In a phase 3 trial in patients infected with HCV genotype 1, treatment-naive patients, including a subgroup with compensated cirrhosis, 89% achieved SVR after 12 weeks of treatment with sofosbuvir and peginterferon or ribavirin.7
In the COmbination of SiMeprevir and sOfoSbuvir in HCV-infected patients (COSMOS) study, we investigated the safety and efficacy of combined oral simeprevir and sofosbuvir, with or without ribavirin, in adults with chronic HCV genotype 1 infections who had previously not responded to or not received standard therapy with peginterferon and ribavirin.
Methods
Study design and patients
This study was a randomised open-label trial done in 23 US centres between Nov 2, 2011, and Jan 29, 2014. We intended to study primarily patients with HCV genotype 1a, as this is a particularly challenging genotype to treat with protease inhibitors. Full eligibility criteria are provided in the study registration information (NCT01466790). Briefly, eligible patients were aged 18 years or older, had chronic infection with HCV genotype 1 and titres of HCV RNA in plasma higher than 10 000 IU/mL, were HIV seronegative, had compensated liver disease, and had glomerular filtration rates of 60 mL/min per 1·73m2 or higher.8Patients were separated into two sequentially recruited cohorts. Those eligible for cohort 1 were required to be previous non-responders to peginterferon and ribavirin and to have no to moderate liver fibrosis (METAVIR score F0-F2), based on a liver biopsy within 3 years of screening (or between screening and day 1 of treatment), and those eligible for cohort 2 were required to be previous non-responders to peginterferon and ribavirin or treatment naive and have severe liver fibrosis (METAVIR score F3-F4), based on a liver biopsy at any time. All patients gave written informed consent. The study was approved by the institutional review boards of all participating institutions or a central institutional review board.
Randomisation and masking
A central, computer-generated randomisation schedule was used with allocation balanced by use of randomly permuted blocks of six (appendix p 1). The next treatment allocation, provided as a unique code, was obtained by an interactive voice-response or web-response system (appendix p 1). Each code was matched with a treatment kit. Randomisation was stratified by HCV genotype 1 subtype (1a vs other) in cohorts 1 and 2, the patient's IL28B genotype in cohort 1, and by treatment history (naive vs non-responder) in cohort 2. As this was an open-label study, all patients and investigators were aware of treatment allocations.
Treatment
Patients were randomly assigned to four treatment groups in a 2:2:1:1 ratio: simeprevir and sofosbuvir with (group 1) or without (group 2) ribavirin for 24 weeks, or simeprevir and sofosbuvir with (group 3) or without (group 4) ribavirin for 12 weeks. After roughly 20% of patients in cohort 1 had reached the planned end of treatment (n=22), a preplanned safety and efficacy analysis was done. 17 (77%) patients had assessable data for SVR 4 weeks after the end of treatment (SVR4). Upon reviewing the results, the data monitoring committee and sponsor agreed to continue with the same four dosing regimens in cohort 2 as in cohort 1.
Simeprevir was taken orally in a 150 mg capsule once daily. Sofosbuvir was taken as two 200 mg tablets once daily in cohort 1 and as one 400 mg tablet daily in cohort 2 after a change in drug formulation. Ribavirin was administered at 1000 mg daily in patients with bodyweight less than 75 kg, or 1200 mg daily in those with bodyweight 75 kg or higher. Patients were followed up for 24 weeks after 24 weeks of treatment or for 36 weeks after 12 weeks of treatment.
Population-based sequencing of HCV NS3/4A and NS5B regions was done on all blood samples collected at baseline. Samples were also collected at study visits on days 1, 2, 3, 4, 7, 14, 21, and 28, and then every 2 weeks until the end of follow-up at week 48 (final sample collected on Jan 29, 2014), but only those for patients who had not achieved SVR 12 weeks after the planned end of treatment (SVR12) were sequenced.
Safety
Safety data were collected from all patients from the time they gave informed consent until the completion of the last study visit. Assessments during treatment included standard laboratory tests (weekly in all patients from baseline up to and including week 4, then every 2 weeks until week 24, at weeks 28 and 32 in groups 1 and 2, and at week 48 in all groups), measurement of vital signs, electrocardiography, and incidence and severity of adverse events (AEs).9 Laboratory value abnormalities and AEs were graded by investigators according to the WHO grading scale.10 Increased bilirubin was deemed to be an event of interest because simeprevir is an inhibitor of the hepatic transporters OATP1B1 and MRP2. Rash (any type), pruritus, anaemia, and neutropenia were deemed events of clinical interest because they have been reported in phase 3 studies of other HCV protease inhibitors combined with peginterferon or ribavirin.11-14
Pharmacokinetics of simeprevir, sofosbuvir, and metabolites of sofosbuvir were measured in all patients at weeks 2, 4, 8, 12, and 24, and in a subgroup of patients at week 2 before and 0·5, 1, 2, 3, 4, 5, 6, 8, 12, and 24 h after the study treatment was taken (appendix p 1).
Statistical analysis
No formal sample size calculation was performed as this was an exploratory study. Nevertheless, we calculated that a sample size of 30 patients in each of the ribavirin groups and 15 in the no ribavirin groups would yield good precision of the 95% CI around the observed response rate (appendix p 1). With enrolment of only non-responders into cohort 1, however, the target number of patients was not reached and, therefore, in cohort 2 inclusion of treatment-naive patients and non-responders was allowed.
The primary endpoint was SVR12 (HCV RNA titres lower than 25 IU/mL). Secondary efficacy endpoints were SVR4 and SVR 24 weeks after the planned end of treatment (SVR24), rapid virological response (HCV RNA undetectable 4 weeks after the start of treatment), on-treatment failure, and viral relapse. Concentrations of HCV RNA in plasma were measured with the COBAS Taqman HCV/HPS v2.0 assay (Roche, Molecular Diagnostics, Pleasanton, CA, USA; lower limit of quantification 25 IU/mL; limit of detection 15 IU/mL).
The primary analysis was done in the intention-to-treat (ITT) population (all randomised patients who received at least one dose of study medication). An additional (post-hoc) analysis of the primary endpoint was done that excluded patients who discontinued treatment prematurely for non-virological reasons or for whom assessment data were missing. Proportional analyses were performed separately for each cohort and, to increase sample size, on pooled cohort data. This trial is registered with ClinicalTrials.gov, number NCT01466790.
Role of the funding source
The funder of the study was responsible for study design, data collection, data analysis, and data interpretation, and helped write and review the report. The investigators were also responsible for data interpretation and writing of the report. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Results
168 patients were enrolled and randomised, and 167 started treatment (n=80 in cohort 1, n=87 in cohort 2; figure 1). Five patients were enrolled despite not satisfying all selection criteria (four in cohort 1 and one in cohort 2), two despite receiving a prohibited concomitant treatment (one in each of cohorts 1 and 2), and one despite missing a key visit at week 36 (cohort 2); as they comprised less than 10% of the overall study population, no per-protocol analysis was done. Except for previous treatment response, Gln80Lys (Q80K) polymorphism, and fibrosis stage, baseline demographics and clinical characteristics were similar across the two cohorts and all treatment groups (table 1). Most patients were white men. Median age was 57 years (range 27-70 years). Most patients were infected with HCV genotype 1a, 45% with the Gln80Lys polymorphism (none in HCV genotype 1b). 23 (14%) patients had the IL28B CC genotype. Ten patients discontinued therapy within 12 weeks of starting study treatment, five [6%] in each cohort, all in the 24-week treatment groups (figure 1). Four withdrawals were due to AEs (table 2).
154 (92%) of 167 of patients in the ITT population achieved SVR12, 90% (95% CI 81-96) in cohort 1 and 94% (87-98) in cohort 2 (figure 2). In separate assessments of cohorts 1 and 2, SVR rates were seen in high proportions of patients with HCV genotype 1a, including in the presence of the HCV Gln80Lys polymorphism, and of those with HCV genotype 1b (figure 2). In cohort 1, rates of virological response were similar in patients with IL28B CC and non-CC genotypes, irrespective of whether they received ribavirin (appendix p 1). The rates of SVR remained high when patients with compensated cirrhosis (METAVIR score F4) were considered separately (appendix p 1), including in the presence of the HCV Gln80Lys polymorphism at baseline (11 [92%] of 12). The results were not significantly altered by use of ribavirin, duration of treatment, or by use of previous treatment (appendix p 1).
Similarities in baseline characteristics and SVR12 rates for cohorts 1 and 2 allowed data pooling to assess the effects of ribavirin, treatment duration, and baseline HCV Gln80Lys status on the primary outcome. In the ITT population, SVR12 was seen in 98 (91%) of 108 patients who received ribavirin versus 56 (95%) of 59 of those who did not. Rates were similar by treatment status (38 [95%] of 40 treatment-naive patients vs 116 [91%] of 127 previous non-responders) or treatment duration (77 [94%] of 82 after 12 weeks of treatment vs 77 [91%] of 85 after 24 weeks). Neither ribavirin nor treatment duration had a clear effect on SVR in patients infected with HCV with the Gln80Lys polymorphism at baseline. Rates were high in patients infected with HCV genotype 1a, irrespective of HCV Gln80Lys polymorphism status at baseline, and in those infected with genotype 1b (figure 2). The only baseline characteristic that notably affected virological response was age 45 years or younger, which was associated with a disproportionate number of non-virological failures (figure 3).
In cohort 1, group 1, 19 (79%) of 24 patients achieved SVR12, which was notably lower than in the other treatment groups (all more than 90%). The difference was due to patients discontinuing treatment for non-virological reasons or missing data at the time of virological response assessment (appendix p 2). When these patients were excluded, the rate in cohort 1, group 1 increased to 95% (19 of 20 patients). The overall rate after exclusion was 96% (appendix p 2). Moreover, after exclusion the response rates in the subgroups of pooled patients were 96% in patients who did (98 of 102) and did not (54 of 56) receive ribavirin, 94% (77 of 82) after 12 weeks of treatment and 99% (75 of 76) after 24 weeks of treatment, and 95% (117 of 123) in patients with HCV genotype 1a (49 [93%] of 53 infected with HCV with the Gln80Lys polymorphism at baseline and 68 [97%] of 70 of those without). Among treatment-naive patients 37 (95%) of 39 achieved SVR12, compared with 115 (97%) of 119 previous non-responders (METAVIR scores F0-F4). SVR rates remained high across all subgroups: cohort 1, 54 (95%) of 57 for genotype 1a (24 [89%] of 27 infected with HCV with the Gln80Lys polymorphism and 30 [100%] of 30 without), four (100%) of four with the IL28B CC genotype, and 67 (96%) of 70 with non-CC genotypes; cohort 2, 63 (96%) of 66 with genotype 1a (including 25 [96%] of 26 with the HCV Gln80Lys polymorphism and 38 [95%] of 40 of those without). All genotype 1b patients after exclusions in both cohorts achieved SVR (35/35).
All patients who achieved SVR12 also achieved SVR4. More than 91% of patients overall achieved SVR4 (appendix p 3). Rapid virological response was achieved in 81% of patients overall (appendix p 3), but SVR12 was still achieved in all but one who had detectable HCV RNA titres 4 weeks after the start of treatment.
No patients experienced on-treatment virological failure, including viral breakthrough. Six patients had viral relapse after the end of treatment (appendix p 4). At the time of relapse, five of the six had developed resistance-associated mutations to simeprevir (Arg155Lys, Asp168Glu, Ile170Thr), but none to sofosbuvir. Five had received 12 weeks of treatment, and four had the HCV Gln80Lys polymorphism at baseline (appendix p 4). Viral relapse was not associated with reduced speed of viral decay during weeks 1-4 of treatment (appendix p 5).
The pharmacokinetics analysis showed no effect of sofosbuvir on simeprevir exposure. Simeprevir increased exposure to sofosbuvir relative to historical data, but did not increase the major circulating metabolite of sofosbuvir (PSI-6202). The observed changes were not clinically important and no dose modifications were required (appendix pp 6-7).
For assessment of safety outcomes, the data from cohorts 1 and 2 were pooled (table 2). Four (2%) patients discontinued all study treatment because of AEs. All were randomised to 24 weeks of therapy but three discontinued before week 12; no patients discontinued ribavirin only. The most common AEs were fatigue in 52 (31%) patients, headache in 33 (20%), and nausea in 26 (16%), but none of these was deemed to be clinically important. 87% (range 71-94) of patients reported at least one AE during the treatment period, but most were grade 1 or 2 (table 2).
One (2%) of 54 patients in each of groups 1 and 3 and three (10%) of 31 in group 2 reported grade 4 AEs, but, overall, grade 3 or 4 AEs were reported in less than 5% of all patients, except increased blood amylase concentration, which was reported in two (4%) of patients in group 1, two (7%) in group 2, and three (6%) in group 3. There were no cases of clinical pancreatitis, and laboratory values improved despite continued dosing.
Rash (any type, grade 1 or 2), pruritus (in the 24-week treatment groups), hyperbilirubinaemia, and anaemia were reported more frequently in the groups receiving ribavirin than in those not receiving ribavirin (table 2, appendix pp 8-9). Most rash and photosensitivity AEs were deemed to be related to one or more of the study drugs but did not lead to permanent discontinuation of therapy and decreased from 4 weeks after the end of treatment.
The most common laboratory parameter abnormalities were hyperglycaemia (up to grade 3), bilirubin, and amylase concentration (both grade 4 in one patient each, appendix pp 9-10). 75 (45%) patients had hyperbilirubinaemia, mostly in the groups receiving ribavirin (group 1, 29 [54%] of 54 and group 3, 32 [59%] of 54). Of these, ten (6%) and one (1%) episodes were grade 3 and 4, respectively. However, few hyperbilirubinaemia events were reported as AEs (table 2). Increases in bilirubin concentrations (mainly indirect) were transient (range of median increases 7·0-10·0 μmol/L from baseline, with the highest values being seen in week 2 and resolving completely after the end of treatment), mostly in patients receiving ribavirin. No concurrent transaminase increases were seen. Most episodes of hyperglycaemia were grade 1 (43%) and most amylase abnormalities were grade 1 or 2 (26% and 14%, respectively).
Haemoglobin concentration lower than 105 g/L was less frequent in patients receiving 24 weeks of treatment without ribavirin than in the other groups (group 2, 3% vs group 1, 17%; group 3, 6%; and group 4, 4%; appendix pp 9-10). Anaemia reported as an AE was also less frequent in the groups not receiving ribavirin than in those receiving ribavirin (table 2).
Serious AEs were infrequent. All were seen in groups 1 and 2 in cohort 2 (two during and two after the first 12 weeks of treatment; group 1 retinal tear and visual impairment n=1, cholelithiasis n=1, and toxicity to alcohol and unprescribed narcotics n=1, and group 2 anaemia n=1). Two patients died during the study, one of ischaemic stroke during follow-up in cohort 1 and one due to a fatal accident (patient with toxicity to alcohol and unprescribed narcotics) during the treatment period. Serious AEs and deaths were all deemed unrelated to treatment.
The frequency of grade 1-2 AEs was similar in the two cohorts; the frequency of grade 3-4 AEs was also similar. Among patients in cohort 1 the rate of grade 3 or 4 AEs was not affected by treatment duration or the addition of ribavirin. In cohort 2, most grade 3 or 4 AEs occurred in group 1 (appendix pp 8-9).
Discussion
The use of combined simeprevir and sofosbuvir in patients infected with HCV genotype 1 led to high rates of SVR12 even though patients had multiple factors traditionally associated with low cure rates with peginterferon and ribavirin-based treatments, including compensated cirrhosis and non-response.
The treatment was generally well tolerated.
Rapid virological response (within 4 weeks of the start of treatment) was seen in 81% of patients. SVR12 rates were similar in patients with and without rapid response (96% vs 97%) and, therefore, rapid response did not predict SVR. Treatment for 24 weeks and the addition of ribavirin did not clearly improve SVR rates in patients with advanced fibrosis or compensated cirrhosis, previous non-response to therapy, or the HCV Gln80Lys polymorphism present at baseline. Viral relapse could not be predicted from viral kinetics during the first 4 weeks of treatment, although, notably, SVR was robust in the 12-week treatment groups (94% when non-virological failures were excluded from the analysis). Five of the six patients with viral relapse were treated for 12 weeks, including all three patients who had advanced fibrosis. A benefit from extending treatment to 24 weeks in at least a subset of patients cannot, therefore, be entirely ruled out. The HCV mutations seen in patients treated with simeprevir and sofosbuvir have also been seen in those who had treatment failure with simeprevir plus peginterferon and ribavirin15 and, either alone or in combination with the Gln80Lys polymorphism, generally have conferred high-level resistance to simeprevir.15
Patients with previous non-response to protease inhibitor therapy were not enrolled in this study, although they have been included in studies involving the NS5A inhibitor, daclatasvir, which has a mechanism of action not affected by protease-inhibitor-related mutations. Similarly, we did not enrol patients with previous failure to NS5A inhibitors. In view of the prevalence of the natural NS5A polymorphism associated with resistance to NS5A inhibitors, however, some patients with pre-existing NS5A mutations might have been successfully treated with combined simeprevir and sofosbuvir.
In phase 3 studies of simeprevir combined with peginterferon and ribavirin, the Gln80Lys polymorphism in HCV genotype 1a was associated with reduced rates of SVR12 (58%) compared with that in patients infected with HCV without this mutation (84%).16 By contrast, in COSMOS, the combination of simeprevir and sofosbuvir resulted in high rates of SVR in patients infected with HCV genotype 1a with the Gln80Lys polymorphism, including in those with compensated cirrhosis (91% in the ITT analysis). Moreover, the efficacy against HCV with the Gln80Lys polymorphism was not affected by whether or not ribavirin was used or the treatment duration. Despite the decreased sensitivity to simeprevir conferred by the Gln80Lys polymorphism,16 its effect on treatment outcomes seemed to be substantially attenuated or even eliminated by the addition of sofosbuvir, which is a potent antiviral agent with a high barrier to resistance. The potency of the combined regimen is underscored by the difference in SVR rates between this study and 10% in a study of non-responders infected with HCV genotype 1 who were treated with sofosbuvir and ribavirin.17 Indeed, the efficacy achieved without ribavirin in our study might avoid ribavirin-associated AEs. Similar high rates of SVR12 were reported in a study of patients infected with HCV genotype 1 who received daclatasvir plus sofosbuvir, with or without ribavirin, for 12 or 24 weeks.18 Other interferon-free regimens are expected to become available, such as ledipasvir (GS-5885) and sofosbuvir, ABT-450 (a protease inhibitor) and ritonavir with ombitasvir and dasabuvir, and asunaprevir and daclatasvir, although no head-to-head studies have yet been done to compare these regimens.
In the COSMOS study, the viral relapse rate was low and mainly coincided with the emergence of mutations that have previously been associated with simeprevir resistance.3-5 No sofosbuvir-related mutations were observed.
In accordance with other studies of simeprevir or sofosbuvir in ribavirin-containing regimens,19 fatigue, headache, and nausea were the most frequent AEs and abnormalities in laboratory parameters correlated with use of ribavirin. AEs in COSMOS were mild to moderate, clinically manageable, and less frequent than are seen with interferon-containing regimens. Few grade 3 or 4 AEs or serious AEs were reported, and very few patients discontinued treatment due to AEs. In particular, METAVIR score was not associated with treatment discontinuation due to AEs, which supports the view that this therapeutic regimen is well tolerated in patients with compensated cirrhosis.
Our results support the hypothesis that the additive effects of ribavirin-induced haemolysis and simeprevir inhibition of transporters involved in the metabolism of bilirubin lead to the increases in bilirubin concentrations seen previously in patients receiving simeprevir-based therapy.19 In our study, raised bilirubin concentrations were almost exclusively seen in patients who received ribavirin, which suggests that the effect of simeprevir on bilirubin metabolism would not be clinically measurable in the absence of ribavirin-induced haemolysis.
Of note, on the basis of interim data from this study,20 clinical guidance from the American Association for the Study of Liver Diseases and the Infectious Disease Society of America21 has been changed to recommend that patients infected with HCV genotype 1 who do not respond to peginterferon or ribavirin or who are treatment-naive and not eligible for peginterferon-based therapy should be treated with 400 mg sofosbuvir and 150 mg simeprevir once daily, with or without ribavirin, for 12 weeks. The European Association for the Study of the Liver guidelines22 state that patients infected with HCV genotype 1 can be treated daily with an interferon-free regimen of 150 mg simeprevir plus 400 mg sofosbuvir for 12 weeks, and that the addition of ribavirin does not confer a major advantage. These guidelines do, however, suggest considering the use of ribavirin in patients with predictors of poor response to therapy, especially previous non-responders, patients with cirrhosis, or both.
The strengths of this study are the enrolment of a population of patients traditionally judged difficult to cure owing to baseline characteristics historically associated with poor rates of SVR, and the evaluation of a short ribavirin-free treatment regimen. Limitations include the open-label study design, a small number of patients per treatment group, and inadequate statistical power to conclusively demonstrate a lack of difference between subgroups.
Overall, the combination of simeprevir and sofosbuvir seemed to be efficacious and well tolerated by previously non-responsive and treatment-naive patients with chronic HCV genotype 1 infections (panel). Efficacy was seen in those with compensated cirrhosis, and ribavirin might not be required to achieve high rates of SVR. This regimen addresses the unmet clinical need for highly effective oral interferon-free treatment options in patients with advanced fibrosis or cirrhosis and who have previously not responded to peginterferon or ribavirin. On the basis of our data, the efficacy and safety of oral combined simeprevir and sofosbuvir without ribavirin is being assessed in the phase 3 OPTIMIST clinical program me.
------------------------
Panel
Research in context
Systematic review
We searched PubMed up to May 1, 2014, for clinical trials done in patients infected with hepatitis C virus (HCV) genotype 1, with the terms "HCV", "hepatitis", and "interferon-free". The HCV treatment landscape has evolved from the well established standard of care with pegylated interferon (peginterferon) and ribavirin to include direct-acting antiviral agents, such as boceprevir and telaprevir. Although these agents in combination with peginterferon and ribavirin have improved sustained virological response rates, they are not suitable for all patients owing to frequent dosing and worsening of anaemia and rash adverse events. In addition, the adverse events associated with injectable interferon-based therapies hinder or prevent their use in some patients. We investigated the efficacy and safety of two second-generation direct-acting antiviral agents, with or without ribavirin.
Interpretation
Our study combined an NS3/4A protease inhibitor and a nucleotide analogue NS5B polymerase inhibitor. The rates of sustained virological response were high and those of virological failure were low. These results were similar to those in a study of combined ledipasvir, an investigational NS5A inhibitor, with sofosbuvir.18 Data from our study19 support the current clinical practice guidelines from the American Association for the Study of Liver Diseases and the Infectious Diseases Society of America,20 which recommend the use of sofosbuvir and simeprevir in patients infected with HCV genotype 1 who are ineligible for interferon-based regimens or have previously not responded to interferon-based therapy. The European Association for the Study of the Liver recommends an interferon-free regimen of daily simeprevir and sofosbuvir, with or without ribavirin, for 12 weeks.
|
|
| |
| |
|
|
|