| |
Sofosbuvir and Velpatasvir for HCV Genotype 1, 2, 4, 5, and 6 Infection
|
| |
| |
Download the PDF here
Download the PDF here
Sofosbuvir and Velpatasvir for HCV Genotype 1, 2, 4, 5, and 6 Infection
Jordan J. Feld, M.D., M.P.H., Ira M. Jacobson, M.D., Christophe Hezode, M.D., Tarik Asselah, M.D., Ph.D., Peter J. Ruane, M.D., Norbert Gruener, M.D., Armand Abergel, M.D., Ph.D., Alessandra Mangia, M.D., Ching-Lung Lai, M.D., Henry L.Y. Chan, M.D., Francesco Mazzotta, M.D., Christophe Moreno, M.D., Ph.D., Eric Yoshida, M.D., Stephen D. Shafran, M.D., William J. Towner, M.D., Tram T. Tran, M.D., John McNally, Ph.D., Anu Osinusi, M.D., Evguenia Svarovskaia, Ph.D., Yanni Zhu, Ph.D., Diana M. Brainard, M.D., John G. McHutchison, M.D., Kosh Agarwal, M.D., and Stefan Zeuzem, M.D. for the ASTRAL-1 Investigators
NEJM November 16, 2015
ABSTRACT
Background
A simple treatment regimen that is effective in a broad range of patients who are chronically infected with the hepatitis C virus (HCV) remains an unmet medical need.
Methods
We conducted a phase 3, double-blind, placebo-controlled study involving untreated and previously treated patients with chronic HCV genotype 1, 2, 4, 5, or 6 infection, including those with compensated cirrhosis. Patients with HCV genotype 1, 2, 4, or 6 were randomly assigned in a 5:1 ratio to receive the nucleotide polymerase inhibitor sofosbuvir and the NS5A inhibitor velpatasvir in a once-daily, fixed-dose combination tablet or matching placebo for 12 weeks. Because of the low prevalence of genotype 5 in the study regions, patients with genotype 5 did not undergo randomization but were assigned to the sofosbuvir-velpatasvir group. The primary end point was a sustained virologic response at 12 weeks after the end of therapy.
Results
Of the 624 patients who received treatment with sofosbuvir-velpatasvir, 34% had HCV genotype 1a, 19% genotype 1b, 17% genotype 2, 19% genotype 4, 6% genotype 5, and 7% genotype 6. A total of 8% of patients were black, 19% had cirrhosis, and 32% had been previously treated for HCV. The rate of sustained virologic response among patients receiving sofosbuvir-velpatasvir was 99% (95% confidence interval, 98 to >99). Two patients receiving sofosbuvir-velpatasvir, both with HCV genotype 1, had a virologic relapse. None of the 116 patients receiving placebo had a sustained virologic response. Serious adverse events were reported in 15 patients (2%) in the sofosbuvir-velpatasvir group and none in the placebo group.
Conclusions
Once-daily sofosbuvir-velpatasvir for 12 weeks provided high rates of sustained virologic response among both previously treated and untreated patients infected with HCV genotype 1, 2, 4, 5, or 6, including those with compensated cirrhosis. (Funded by Gilead Sciences; ClinicalTrials.gov number, NCT02201940.)
The hepatitis C virus (HCV), a single-stranded RNA virus of the family Flaviviridae with six major genotypes, infects up to 150 million people worldwide.1,2 Chronic HCV infection causes progressive liver fibrosis, which can lead to cirrhosis, hepatic decompensation, and hepatocellular carcinoma.3,4 As many as half a million people die annually from liver disease associated with chronic HCV infection.5
In recent years, the development of drugs that directly interfere with HCV replication has revolutionized HCV treatment. There are now effective combinations of direct-acting antiviral agents for most patients, but in choosing an appropriate regimen, clinicians must take into account the patient's treatment history, HCV genotype and subtype, stage of fibrosis, and, in some cases, patterns of antiviral resistance. Some regimens also include ribavirin, which has known hematologic and other side effects and is teratogenic.6,7 The development of a ribavirin-free single-tablet regimen of short duration that is effective in a broad range of patients with HCV infection would simplify clinical decision making and reduce the need for pretreatment testing and monitoring during therapy.8
Sofosbuvir is a nucleotide analogue inhibitor of the HCV NS5B polymerase approved for the treatment of HCV in combination with a variety of other agents, including NS5A inhibitors, ribavirin, and peginterferon-ribavirin. Velpatasvir (formerly GS-5816, Gilead Sciences) is a new pangenotypic HCV NS5A inhibitor with antiviral activity against HCV replicons in genotypes 1 through 6.9-11 In phase 2 trials, the combination of 400 mg of sofosbuvir and 100 mg of velpatasvir with or without ribavirin resulted in high rates of sustained virologic response in a broad range of patients with HCV. These included previously treated and untreated patients, those with and without compensated cirrhosis, and those infected with HCV of all six genotypes.12,13 The inclusion of ribavirin did not appear to improve efficacy but was associated with a slightly increased incidence of some adverse events, including hematologic abnormalities.
We conducted a phase 3 trial (ASTRAL-1) to assess the efficacy and safety of 12 weeks of treatment with a fixed-dose combination of velpatasvir and sofosbuvir among both previously treated and untreated patients who were chronically infected with HCV genotype 1, 2, 4, 5, or 6, including those with compensated cirrhosis.
Methods
Patients
We enrolled patients at 81 sites in the United States, Canada, Europe, and Hong Kong from July 18, 2014, through December 19, 2014. Eligible patients were 18 years of age or older who had chronic infection with HCV genotype 1, 2, 4, 5, or 6. All patients provided written informed consent.
The original clinical-development program for sofosbuvir-velpatasvir involved two trials - one in patients with HCV genotype 1, 2, 4, 5, or 6 (ASTRAL-1) and one in patients with HCV genotype 3. A separate trial with an active comparator group was deemed to be necessary for patients with HCV genotype 3 in light of the special clinical challenges presented in this population. After the protocol for the present study was finalized and trial activity had begun, the Food and Drug Administration requested a separate trial with an active comparator for patients with HCV genotype 2. Because enrollment in the present study had already begun, we did not amend the protocol to exclude patients with HCV genotype 2. Therefore, two additional phase 3 trials were conducted to evaluate sofosbuvir-velpatasvir in patients with HCV genotype 2 (ASTRAL-2) and HCV genotype 3 (ASTRAL-3), and the results are reported now in the Journal.14,15
The protocol targeted an enrollment of approximately 20% of patients who had been previously treated for HCV with a regimen containing interferon and who had not had a sustained virologic response. Those who had discontinued previous HCV treatment because of an adverse event were not eligible. Patients who had previously received any nucleotide analogue HCV NS5B inhibitor or any NS5A inhibitor were not eligible. Approximately 20% of patients could have evidence of cirrhosis, which was defined as liver-biopsy results showing a Metavir fibrosis score of 4 or an Ishak score of 5 or more, a FibroTest score of more than 0.75 and a ratio of aspartate aminotransferase to platelets of more than 2, or a FibroScan reading of more than 12.5 kPa. There were no upper limits for age or body-mass index. Patients with a history of hepatic decompensation or hepatocellular carcinoma were not eligible for enrollment. Full eligibility criteria are provided in the protocol, available with the full text of this article at NEJM.org.
Study Design
In this multicenter, double-blind, placebo-controlled trial, patients with HCV genotype 1, 2, 4, or 6 were randomly assigned in a 5:1 ratio to receive a fixed-dose combination tablet containing 400 mg of sofosbuvir and 100 mg of velpatasvir, administered orally once daily for 12 weeks, or a placebo tablet to match the active treatment once daily for 12 weeks. Patients in the placebo group were eligible for deferred treatment with 12 weeks of sofosbuvir-velpatasvir. Randomization was stratified according to genotype (1, 2, 4, 6, or indeterminate) and the presence or absence of cirrhosis. Given the low prevalence of genotype 5 HCV infection in the regions in which the study was conducted, we targeted the enrollment of only 20 patients with HCV genotype 5. These patients did not undergo randomization but were enrolled in the sofosbuvir-velpatasvir group only.
Study Assessments
Serum HCV RNA was measured by means of the COBAS AmpliPrep/COBAS TaqMan HCV Quantitative Test, version 2.0 (Roche Molecular Systems), with a lower limit of quantification of 15 IU per milliliter. HCV genotype and subtype were determined with the use of the VERSANT HCV Genotype INNO-LiPA 2.0 assay (Siemens). IL28B genotyping was performed by means of polymerase-chain-reaction amplification and sequencing of the rs12979860 single-nucleotide polymorphism.
Assessments during treatment included standard laboratory testing, serum HCV RNA, vital signs, electrocardiography, and symptom-directed physical examinations. All adverse events were recorded and graded according to a standardized scale. (Details are provided in the study protocol.)
Deep sequencing of the target regions of velpatasvir and sofosbuvir, HCV NS5A and NS5B, respectively, was performed for all patients at baseline and again for all patients with virologic failure in samples obtained at the time of failure. The sequences from baseline samples were compared with those obtained at the time of virologic failure to detect emergent resistance-associated variants. Resistance-associated variants that were present in more than 1% of sequence reads are reported.
End Points
The primary efficacy end point was the rate of sustained virologic response, which was defined as an HCV RNA level of less than 15 IU per milliliter at 12 weeks after the end of treatment in all patients who received at least one dose of sofosbuvir-velpatasvir or placebo. Secondary end points included the rate of adverse events and treatment discontinuations because of adverse events.
Study Oversight
This study was approved by the institutional review board or independent ethics committee at each participating study site and was conducted in compliance with the Declaration of Helsinki, Good Clinical Practice guidelines, and local regulatory requirements. The study was designed and conducted by the sponsor (Gilead Sciences) in collaboration with the principal investigators. The sponsor collected the data, monitored study conduct, and performed the statistical analyses. An independent data and safety monitoring committee reviewed the progress and oversight of the study. The investigators, participating institutions, and sponsor agreed to maintain confidentiality of the data. All the authors had access to the data and assume responsibility for the integrity and completeness of the reported data and fidelity to the protocol. The initial draft of the manuscript was prepared by a professional writer employed by Gilead Sciences and the first and last authors with input from all the authors.
Statistical Analysis
The primary efficacy analysis was designed to test for the superiority of the rate of sustained virologic response among patients receiving sofosbuvir-velpatasvir to a prespecified performance goal of 85% by means of a two-sided exact one-sample binomial test. This 85% rate was not a historical control derived from rates of sustained virologic response in prior HCV treatment trials, since it would not be possible to calculate a single historical rate for the different standard treatments recommended for the various genotypes included in this study. Rather, it is a benchmark rate that is based on the general trend toward increasing rates of sustained virologic response in recent years and the general appeal of using a fixed, clinically relevant threshold as a measure of treatment benefit.16 We determined that the enrollment of 500 patients in the sofosbuvir-velpatasvir group would provide a power of 90% to detect an improvement of at least 5 percentage points in the rate of sustained virologic response over the performance goal of 85%, on the basis of the two-sided exact one-sample binomial test at the 0.05 significance level. We used the Clopper-Pearson method to calculate point estimates and two-sided 95% exact confidence intervals for rates of sustained virologic response for the sofosbuvir-velpatasvir group as a whole, as well as according to HCV genotype (1a, 1b, 2, 4, 5, or 6), and various subgroups.
The inclusion of a placebo group was designed to provide the basis for evaluating the safety profile of sofosbuvir-velpatasvir in a population with expected coexisting medical conditions. We used a double-blind approach in the study-group assignments to ensure the elimination of bias in assessments of safety. No formal comparison of safety between the groups was planned.
Results
Baseline Characteristics
Of the 847 patients who were initially screened, 741 were enrolled and 706 underwent randomization; the 35 patients with genotype 5 infection were enrolled in the sofosbuvir-velpatasvir group, as prespecified (Table S1 and Fig. S1 in the Supplementary Appendix, available at NEJM.org). One patient, who was lost to follow-up after undergoing randomization to the sofosbuvir-velpatasvir group but before receiving treatment, was excluded from the safety and efficacy analyses.
The demographic and baseline clinical characteristics of the 116 patients receiving placebo and the 624 patients receiving sofosbuvir-velpatasvir were generally balanced (Table 1). In the sofosbuvir-velpatasvir group, 34% of the patients had HCV genotype 1a, 19% genotype 1b, 17% genotype 2, 19% genotype 4, 6% genotype 5, and 7% genotype 6. Most patients were white (79%) and male (60%). Nineteen percent of the patients had cirrhosis, 69% had a non-CC IL28B genotype (which has been associated with a reduced response to HCV treatment), and 32% had received previous unsuccessful treatment for HCV. Of the 201 patients in the sofosbuvir-velpatasvir group who had received previous treatment, 28% had received a regimen of peginterferon, ribavirin, and a protease inhibitor, and 61% had received peginterferon and ribavirin; 48% of these patients had persistently detectable HCV RNA while receiving previous treatment, and 51% had a virologic relapse or breakthrough. A total of 51% of patients were enrolled in Europe, 46% in North America (Canada and the United States), and 3% in Hong Kong.
Efficacy
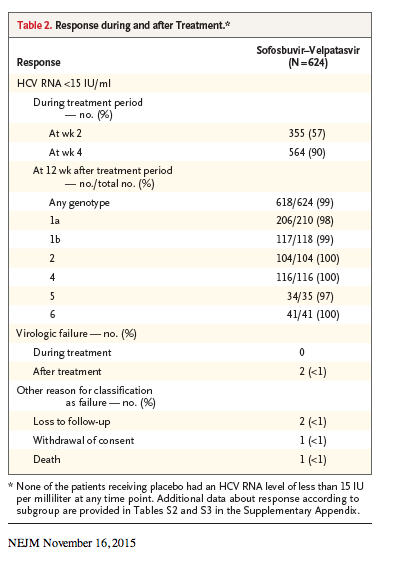
Overall, the rate of sustained virologic response among patients who received 12 weeks of sofosbuvir-velpatasvir was 99% (95% confidence interval [CI], 98 to >99), which was significantly superior to the prespecified performance goal of 85% (P<0.001) (Table 2). None of the 116 patients in the placebo group had a sustained virologic response.
Rates of sustained virologic response were similar regardless of the HCV genotype: 98% (95% CI, 95 to >99) in patients with genotype 1a infection, 99% (95% CI, 95 to 100) with genotype 1b, 100% (95% CI, 97 to 100) with genotype 2, 100% (95% CI, 97 to 100) with genotype 4, 97% (95% CI, 85 to >99) with genotype 5, and 100% (95% CI, 91 to 100) with genotype 6. Of the 121 patients with any genotype who had cirrhosis, 120 (99% [95% CI, 95 to >99]) had a sustained virologic response.
Of the 624 patients who received at least one dose of sofosbuvir-velpatasvir, 2 (<1%) had virologic failure: a 56-year-old white man without cirrhosis who had received no previous treatment for genotype 1a HCV infection and a 58-year-old black man with cirrhosis who had persistently detectable HCV RNA during previous peginterferon-ribavirin treatment for genotype 1b HCV infection. The 2 men had undetectable serum HCV RNA at week 4 of treatment, and both had a virologic relapse by post-treatment week 4. Further details concerning these 2 patients are provided in Table S4 in the Supplementary Appendix.
Four other patients in the sofosbuvir-velpatasvir group are not counted as having had a sustained virologic response. Two of the four were lost to follow-up (one did not return after completing 45 days of treatment; the other completed treatment and had undetectable serum HCV RNA at post-treatment week 4 but did not return for the post-treatment week 12 visit), one discontinued treatment because of an adverse event, and one died during follow-up. (Details regarding the last two patients are provided in the Safety subsection.)
Rates of sustained virologic response in all patient subgroups, including those with cirrhosis (99%) and prior treatment experience (>99%), were high (Tables S2 and S3 in the Supplementary Appendix).
Viral Resistance Testing
At baseline, NS5A resistance-associated variants were detected in 257 of 616 patients (42%) for whom sequencing data were available. Of these 257 patients, 255 (99%) had a sustained virologic response. The 2 patients who had virologic failure had NS5A-resistant variants at baseline and at the time of relapse. The patient with HCV genotype 1a infection who had a relapse had the Q30R variant in 2.6% of the viral population at baseline. At time of relapse, the Q30R variant was no longer present, but the Y93N variant was detected in more than 99% of the viral population. The second patient (with HCV genotype 1b who had a relapse) had the Q30L variant (in 1.1% of the viral population), Q30R (in 98.7%), and L31M (in >99%) at baseline and Q30R (in >99%), L31M (in >99%), and Y93H (in 99%) at the time of relapse. The Q30R variant confers an increase by a factor of 2.2 in the 50% effective concentration (EC50) of velpatasvir in the HCV genotype 1a replicon. Arginine (R) variants at position 30 of the NS5A protein were present at baseline in 62 patients in the entire study population: 5 patients with genotype 1, 5 with genotype 2, 50 with genotype 4, and 2 with genotype 5. Of these 62 patients, 60 (97%) had a sustained virologic response.
Variants associated with resistance to NS5B nucleoside inhibitors were detected at baseline in 54 of the 601 patients (9%) for whom sequencing data were available. No S282 variants were detected. All 54 patients had a sustained virologic response.
Safety
Of the 624 patients in the sofosbuvir-velpatasvir group, 1 (<1%) discontinued treatment prematurely because of an adverse event. This patient, a 52-year-old white woman with genotype 1a HCV infection without cirrhosis, discontinued treatment because of an anxiety attack on the 13th day of treatment. Of the 116 patients in the placebo group, 2 (2%) discontinued treatment because of an elevated aminotransferase level, a prespecified criterion for discontinuation.
A total of 15 patients (2%) in the sofosbuvir-velpatasvir group had 19 serious adverse events (Table 3, and Table S5 in the Supplementary Appendix). No single serious adverse event occurred in more than 1 patient. There was one death in the sofosbuvir-velpatasvir group. This patient, a 55-year-old white man with HCV genotype 5a without cirrhosis who had a history of dyslipidemia for which he was taking ezetimibe-simvastatin, died during sleep 8 days after the completion of treatment. The cause of death was not determined. The patient was not taking amiodarone. None of the patients in the placebo group had a serious adverse event.
There was no significant difference in the rates of any adverse event in the sofosbuvir-velpatasvir group and the placebo group (78% and 77%, respectively). The rates of individual adverse events did not differ significantly between the two groups. The most common adverse events were headache, fatigue, nasopharyngitis, and nausea.
Hematologic abnormalities were infrequent in the sofosbuvir-velpatasvir group, affecting 1% of patients or less. No patients in the placebo group had hematologic abnormalities (Table 3). No patient in either study group had a grade 3 or 4 elevation in creatinine (>3.0 mg per deciliter [265 μmol per liter) or total bilirubin (>2.5 mg per deciliter [43 μmol per liter]).
Discussion
Of the 170 million patients who are chronically infected with HCV worldwide, approximately half have HCV genotypes other than genotype 1, including about one third of patients with HCV in the United States.17 Currently approved regimens of direct-acting antiviral agents are not equally effective across all genotypes, which means that testing to determine genotype and subtype is required before treatment can be initiated.6,7 A single combination regimen that is effective in all patients regardless of HCV genotype would obviate the need for pretreatment testing, which is an obstacle to treatment in resource-limited settings and may limit treatment uptake outside of specialty clinics.18
In this international, randomized, double-blind, placebo-controlled phase 3 study, treatment with sofosbuvir-velpatasvir for 12 weeks resulted in high rates of sustained virologic response in patients with HCV genotype 1, 2, 4, 5, or 6, including those with cirrhosis and those who had received previous treatment and those who had not been treated. Virologic failure was rare in patients infected with HCV genotype 1, and there were no virologic failures among those with HCV genotype 2, 4, 5, or 6. The study patients, who were enrolled at 81 sites in eight countries, were diverse with respect to demographic and baseline characteristics. Patients with characteristics that are historically associated with a lower response to treatment - the presence of cirrhosis, prior treatment failure, black race, and non-CC genotype of IL28B - had rates of virologic response similar to those with historically favorable characteristics.
The rate of sustained virologic response we observed in patients with HCV genotype 2 (100%) was similar to that seen in a companion phase 3 trial14 reported in the Journal, in which 99% of the patients with HCV genotype 2 in the sofosbuvir-velpatasvir group had a sustained virologic response. In a companion phase 3 trial reported in the same Journal article, 95% of the patients with HCV genotype 3 who received sofosbuvir-velpatasvir had a sustained virologic response, including 98% of previously untreated patients without cirrhosis, 93% of previously untreated patients with cirrhosis, 91% of previously treated patients without cirrhosis, and 89% of previously treated patients with cirrhosis.
The presence of baseline resistance-associated variants was not associated with virologic failure, which was represented in our study by two relapses and no virologic breakthrough. Although the two patients who had a relapse had resistance-associated variants at baseline and at the time of virologic failure, 99% of the patients with baseline NS5A resistance-associated variants had a sustained virologic response, which suggests that pretreatment testing for resistance-associated variants is probably of little clinical value with sofosbuvir-velpatasvir.
Serious adverse events occurred in 2% of patients in the sofosbuvir-velpatasvir group and in no patients in the placebo group. No single serious adverse event occurred in more than one patient who received sofosbuvir-velpatasvir. The type, frequency, and severity of nonserious adverse events were generally similar in the two study groups. Hematologic abnormalities occurred infrequently in patients receiving sofosbuvir-velpatasvir (≤1% of patients). No patients receiving placebo had hematologic abnormalities.
A factor limiting the generalizability of our results is that we did not enroll patients in certain regions where the less common HCV genotypes and subtypes are highly prevalent. In addition, we excluded patients with HCV genotype 3 and those with decompensated cirrhosis, but these patients are being evaluated in parallel phase 3 studies14,15 that are reported now in the Journal. Also, patients who had previous treatment failure with sofosbuvir or an NS5A-containing regimen were excluded from the study.
In conclusion, treatment with the single-tablet regimen of sofosbuvir-velpatasvir for 12 weeks was highly effective for a broad range of patients with HCV genotype 1, 2, 4, 5, or 6 infection. The treatment was also effective among patients with compensated cirrhosis.
|
|
| |
| |
|
|
|