| |
Cabotegravir plus rilpivirine, once a day, after induction with cabotegravir plus nucleoside reverse transcriptase inhibitors in antiretroviral-naive adults with HIV-1 infection (LATTE): a randomised, phase 2b, dose-ranging trial
|
| |
| |
Download the PDF
Download the PDF
"For patients with HIV-1 RNA concentrations of fewer than 100 000 copies per mL at baseline, of those receiving cabotegravir, 43 (88%) of 49 in the 60 mg group, 40 (75%) of 53 in the 30 mg group, and 37 (71%) of 52 in the 10 mg group had sustained viral suppression after 72 weeks of maintenance therapy (week 96), compared with 32 (59%) of 54 patients receiving efavirenz."
-------------------
Cabotegravir plus rilpivirine, once a day, after induction with cabotegravir plus nucleoside reverse transcriptase inhibitors in antiretroviral-naive adults with HIV-1 infection (LATTE): a randomised, phase 2b, dose-ranging trial
Lancet Infect Dis July 2015
David A Margolis, Cynthia C Brinson, Graham H R Smith, Jerome de Vente, Debbie P Hagins, Joseph J Eron, Sandy K Griffi th, Marty H St Clair,
Marita C Stevens, Peter E Williams, Susan L Ford, Britt S Stancil, Melinda M Bomar, Krischan J Hudson, Kimberly Y Smith, William R Spreen, for the LAI116482 Study Team
Summary
Background
In phase 1 trials, the HIV-1 integrase strand transfer inhibitor cabotegravir (GSK1265744) was well tolerated, both alone, and in combination with the non-nucleoside reverse transcriptase inhibitor rilpivirine. We assessed cabotegravir plus rilpivirine, as a two-drug oral antiretroviral regimen, for the maintenance of viral suppression in antiretroviral-naive HIV-1-infected individuals.
Methods
In the phase 2b Long-Acting antireTroviral Treatment Enabling (LATTE) trial, a multicentre study done in Canada and the USA, antiretroviral-naive HIV-1-infected adults (aged ≥18 years) were randomly allocated in a 1:1:1:1 ratio to oral cabotegravir 10 mg once a day, 30 mg once a day, 60 mg once a day, or oral efavirenz 600 mg once a day with dual nucleoside reverse transcriptase inhibitors (NRTIs) for 24 weeks of induction.
Patients who were virologically suppressed by week 24 received a two-drug maintenance regimen consisting of their randomly allocated cabotegravir dose plus oral rilpivirine 25 mg or continued efavirenz plus NRTIs for an additional 72 weeks.
Patients and investigators were masked to doses of cabotegravir received for 96 weeks, but not to the assignment of cabotegravir or efavirenz. The primary endpoint was the proportion of patients with fewer than 50 copies per mL of HIV-1 RNA (US Food and Drug Administration snapshot algorithm) at week 48. Analysis was by intention to treat. This study is registered with ClinicalTrials.gov, NCT01641809.
Findings
Of 243 patients randomly allocated and treated, 156 (86%) of 181 patients in the cabotegravir groups (52 [87%] of 60, 51 [85%] of 60, and 53 [87%] of 61 patients in the 10 mg, 30 mg, and 60 mg groups, respectively) and 46 (74%) of 62 in the efavirenz group had fewer than 50 copies per mL of HIV-1 RNA after induction therapy.
After patients in the cabotegravir groups were changed over from dual NRTIs to rilpivirine at week 24, 149 (82%; 95% CI 77-88) patients in the cabotegravir groups (48 [80%; 70-90], 48 [80%; 70-90], and 53 [87%; 78-95] patients in the 10 mg, 30 mg, and 60 mg groups, respectively) versus 44 (71%; 60-82) in the efavirenz group were virologically suppressed at week 48, and 137 (76%; 69-82) receiving cabotegravir (41 [68%; 57-80], 45 [75%; 64-86], and 51 [84%; 74-93] patients in the 10 mg, 30 mg, and 60 mg groups, respectively) versus 39 (63%; 51-75) in the efavirenz group were virologically suppressed at week 96.
Treatment-related adverse events were reported by 93 (51%) cabotegravir-treated patients (28 [47%], 32 [53%], and 33 [54%] patients in the 10 mg, 30 mg, and 60 mg groups, respectively) and 42 (68%) efavirenz-treated patients. Six (3%) patients in the cabotegravir groups (one [2%], one [2%], and four [7%] patients in the 10 mg, 30 mg, and 60 mg groups, respectively) withdrew because of treatment-emergent adverse events compared with nine (15%) in the efavirenz group.
Interpretation
Cabotegravir plus dual NRTI therapy had potent antiviral activity during the induction phase. As a two-drug maintenance therapy, cabotegravir plus rilpivirine provided antiviral activity similar to efavirenz plus dual NRTIs until the end of week 96. Combined efficacy and safety results lend support to our selection of oral cabotegravir 30 mg once a day for further assessment. LATTE precedes studies of the assessment of longacting injectable formulations of both drugs as a two-drug regimen for the treatment of HIV-1 infection.
Funding
ViiV Healthcare and Janssen Research and Development.
Introduction
Over the past two decades, novel drugs for HIV-1 antiretroviral therapy have improved long-term viral suppression.1 The discovery of integrase strand transfer inhibitors (INIs) has provided important treatment options for patients with HIV/AIDS. Approved first-generation INIs (raltegravir and elvitegravir) are effective and generally well tolerated.2, 3, 4, 5 Clinical resistance to these first-generation INIs has, however, been reported in treatment-naive and previously treated patients.6, 7, 8, 9, 10, 11, 12 Dolutegravir is a second-generation INI with an improved resistance and administration profile.13 These advances might be further augmented by the development of longacting injectable antiretroviral drugs and regimens with the potential to improve adherence and thereby increase the options for treatment or prophylaxis.
Research in context
Evidence before this study
We searched PubMed, with combinations of the search terms "integrase strand transfer inhibitor" (INI), "integrase inhibitor", "non-nucleoside reverse transcriptase inhibitor", "raltegravir", "dolutegravir", "elvitegravir", "rilpivirine", "two drug HIV therapy", "long acting", and "HIV pre-exposure prophylaxis", with no restrictions on language or publication date. Several small retrospective and uncontrolled studies using INIs plus non-nucleoside reverse transcriptase inhibitors (NNRTIs) as dual HIV-1 therapy have been reported. Results from these studies suggested long-term viral suppression from well tolerated regimens, which warranted further investigation and confirmation in larger randomised controlled studies. Longacting parenteral versions of cabotegravir and rilpivirine are being developed with promising preclinical and clinical pharmacology profiles.
Added value of this study
LATTE 24-week induction results confirm rapid and robust viral suppression with cabotegravir plus dual nucleoside reverse transcriptase inhibitors (NRTIs), a profile that has been well established for other drugs within the INI class. The two-drug maintenance regimen of cabotegravir plus rilpivirine showed viral suppression for an additional 72 weeks similar to the three-drug regimen of efavirenz plus dual NRTIs with similar safety and tolerability and an impressive resistance profile. Results from the LATTE study confirm, for the first time within a randomised controlled trial, the safety and durability of a two-drug INI plus NNRTI regimen after induction therapy in an antiretroviral-naive adult population.
Implication of all the available evidence
Current three-drug combination antiretroviral therapy regimens might result in treatment modification or interruption because of side-effects, toxicities, or complicated administration profiles. New HIV-1 antiretroviral agents need to be developed with a focus on improved safety, efficacy, and resistance profiles, and more convenient administration to enable patient adherence. In view of the potential for long-term side-effects of dual NRTIs and pharmacologically boosted protease-inhibitor regimens, a well tolerated, simplified two-drug therapy that offers sustained viral suppression while avoiding these classes might be particularly desirable within the ageing population with HIV infection and in patients with comorbidities, including renal and cardiovascular disease. Results from LATTE are fundamental for further assessment of parenteral formulations of cabotegravir and rilpivirine towards the increase in treatment options for patients and health-care providers.
Cabotegravir (GSK1265744) is an INI and structural analogue of dolutegravir with potent anti-HIV-1 activity, a half-life of about 40 h when dosed orally, and a low propensity for drug interactions.14 Oral doses of cabotegravir have been generally well tolerated in early trials of healthy and HIV-1-infected individuals.15 Rilpivirine is a non-nucleoside reverse transcriptase inhibitor (NNRTI) with in-vitro activity against HIV-1 wild type and resistant to some NNRTIs.16 Oral rilpivirine at 25 mg once a day has been approved for use in antiretroviral-therapy-naive patients and in some virologically suppressed patients as a replacement antiretroviral treatment regimen. No significant pharmacokinetic interactions occurred between oral cabotegravir and oral rilpivirine,14 lending support to the investigation of a two-drug regimen for the maintenance of viral suppression. Parenteral longacting formulations of cabotegravir and rilpivirine are in clinical development with the goal to establish the safety and efficacy of a two-drug injectable regimen with a dosing interval of at least 4 weeks. So far, both longacting formulations have been generally well tolerated, alone and together, with longlasting plasma exposures after intramuscular injection.17, 18, 19
In the 24-week induction phase of Long-Acting antireTroviral Treatment Enabling (LATTE), a phase 2b study, we assessed viral suppression (HIV-1 RNA <50 copies per mL) by oral cabotegravir plus nucleoside reverse transcriptase inhibitors (NRTIs) compared with efavirenz plus NRTIs. In the maintenance phase of LATTE, we assessed a two-drug oral antiretroviral treatment regimen (cabotegravir plus rilpivirine) for maintenance of viral suppression for an additional 72 weeks compared with a three-drug efavirenz-based antiretroviral therapy.
Methods
Study design and participants
LATTE is a phase 2b, randomised, multicentre, parallel-group, partly masked induction and maintenance study in HIV-1 infected antiretroviral-therapy-naive adults at 49 sites in Canada and the USA. Eligible patients (aged ≥18 years) had HIV-1 infection with screening plasma HIV-1 RNA copies of at least 1000 per mL, had a CD4 cell count of at least 200 per μL, were antiretroviral treatment naive (�10 days of previous treatment), and had no major drug-resistance-associated mutations.20 Exclusion criteria included active US Centers for Disease Control and Prevention category C disease (ie, patients were affected by one or more serious complications or infections associated with late-stage HIV and AIDS), laboratory values of clinical concern, pregnancy, moderate or severe hepatic impairment, clinically relevant hepatitis, anticipated need for hepatitis C treatment, creatinine clearance of less than 50 mL/min, treatment with an HIV-1 vaccine, or treatment with an immunomodulator drug within 90 days of screening. Patients could receive abacavir after screening negative for the HLA-B*5701 allele.
All participating centres obtained ethics committee approval in accordance with the principles of the 2008 Declaration of Helsinki. Each patient gave written informed consent before undergoing study procedures.
Randomisation and masking
Patients were randomly assigned in a 1:1:1:1 ratio to receive oral cabotegravir 10 mg once a day, 30 mg once a day, or 60 mg once a day, or oral efavirenz 600 mg once a day, each with investigator-selected background NRTI (abacavir-lamivudine or tenofovir-emtricitabine fixed-dose combination tablets). Central randomisation, including stratification by screening plasma HIV-1 RNA (<100 000 copies per mL or ≥100 000 copies per mL) and by the use of abacavir-lamivudine or tenofovir-emtricitabine fixed-dose combination tablets as initial background dual NRTIs, was generated through validated randomisation software RandAll (version 2.10). Patients and investigators were masked to doses of cabotegravir received until the end of week 96, but not to the assignment of cabotegravir or efavirenz.
Procedures
In the induction phase, cabotegravir doses with NRTIs were assessed for antiviral activity, safety, and pharmacokinetics over 24 weeks relative to efavirenz with NRTIs. Patients receiving cabotegravir who completed 24 weeks and had viral suppression (plasma HIV-1 RNA <50 copies per mL immediately before week 24) and those in the efavirenz group with a week 24 visit, irrespective of HIV-1 RNA copies of less than 50 per mL, were eligible for the maintenance phase of this study. During maintenance, background NRTIs were discontinued from the study in the cabotegravir groups and the antiretroviral therapy regimen was reduced to the randomly allocated cabotegravir dose in combination with rilpivirine 25 mg once a day for an additional 72 weeks. Patients in the efavirenz group continued background NRTIs until the end of week 96.
Clinical and laboratory analyses were done at baseline, weeks 2, 4, 8, 12, 16, 20, 24, 26, 28, 32, 36, 40, and 48, and every 12 weeks thereafter. Central laboratory facilities (Quest Diagnostics, Valencia, CA, USA) provided genotype, haematology, clinical chemistry, urinalysis, CD4 cell counts, and plasma HIV-1 RNA testing (RealTime HIV-1 PCR assay, Abbott Molecular, Des Plaines, IL, USA). Monogram Biosciences (San Francisco, CA, USA) provided genotype and phenotype profiles. Covance Laboratories (Sample Management, Madison, WI, USA) and PRA Bioanalytical Laboratory (Assen, Netherlands) analysed plasma samples for cabotegravir and rilpivirine concentrations, respectively.
Outcomes
The primary endpoint was the proportion of patients with HIV-1 RNA copies of fewer than 50 per mL at week 48, using the US Food and Drug Administration (FDA) snapshot algorithm.21
Secondary endpoints were the proportion of patients with plasma HIV-1 RNA of less than 50 copies per mL over time; absolute values and change from baseline in plasma HIV-1 RNA and CD4 cell counts; treatment-emergent genotypic or phenotypic resistance to cabotegravir, rilpivirine, and other on-study antiretroviral treatment for protocol-defined virological failures; incidence and severity of adverse events; and laboratory abnormalities. Suspected protocol-defined virological failures were confirmed with a first and a repeat plasma HIV-1 RNA measurement 2-4 weeks apart. A virological non-response was defined as a reduction of less than 1·0 log10 copies per mL in plasma HIV-1 RNA by week 4, or two consecutive plasma HIV-1 RNA copies of at least 200 per mL after week 16. Virological rebound was indicated by at least 200 copies per mL of HIV-1 RNA after previous suppression to less than 200 copies per mL, or two consecutive plasma HIV-1 RNA measurements that showed an increase of greater than 0·5 log10 copies per mL in plasma HIV-1 RNA from the nadir value on study, with the lowest HIV-1 RNA value of at least 200 copies per mL. Patients confirmed to have met the definition of protocol-defined virological failures were discontinued from the study. Adverse events were graded according to the Division of AIDS toxicity scales.22 Cabotegravir dose selection for further assessment in future studies was the primary objective in the LATTE trial, and was done at week 48 (with additional confirmation at week 72); it was based on the results of an analysis of antiviral activity and tolerability with immunological, safety, viral resistance, and pharmacokinetic measurements.
Statistical analysis
The efficacy analysis of the primary endpoint was done at week 48 in the intention-to-treat exposed population (patients who had received at least one dose of study medication); the proportions of patients with plasma HIV-1 RNA of fewer than 50 copies per mL with the FDA snapshot algorithm were summarised by treatment group. The target sample size of 50 patients per group ensured high probability that the correct dose of cabotegravir was selected for further study and allowed for the formal consideration of other factors in dose selection if efficacy was similar between the dose groups. The decision criterion for efficacy was a difference of more than 8% between the cabotegravir dose groups. With 50 patients per dose group, and assuming true response rates of 85% and 75%, respectively, in two dose groups, there was at least 55% chance that the superior dose would be selected while the chance of incorrectly selecting the inferior dose was less than 1%. The chance of the difference in efficacy rates lying between -8% and 8% was 44%.
Data were also summarised for the intention-to-treat maintenance-exposed population, which consisted of any patient who had received at least one dose of study drug during the maintenance phase. Secondary efficacy analyses were observed values and change from baseline in plasma HIV-1 RNA (log10 copies per mL), CD4 cell counts, and time to viral suppression or failure, or both. An internal safety review committee assessed efficacy, safety, and tolerability of cabotegravir at weeks 16 and 24. Safety and tolerability of cabotegravir were compared with efavirenz in terms of incidences of adverse events, serious adverse events, graded laboratory toxicities, summaries of laboratory tests, and vital signs until week 96.
Phenotypes and genotypes of reverse transcriptase, protease, and integrase genes were analysed at baseline and time of suspected protocol-defined virological failures for treatment-emergent mutations using the first sample obtained for suspected virological failure. Cabotegravir pharmacokinetic parameters including the area under the plasma concentration-time curve from time zero to time t, maximum plasma drug concentration, trough concentration (C0), plasma drug concentration at a specified time t after drug administration, time to maximum plasma concentration after drug administration, and total clearance of the drug were estimated from samples obtained before or after dosing at weeks 2, 12, 26, and 36. Rilpivirine pharmacokinetic parameters were estimated from samples obtained before or after dosing at weeks 26 and 36. Pharmacokinetic parameters were calculated by standard non-compartmental analysis with Phoenix WinNonlin Pro (version 5.2 or higher).
Role of the funding source
The funders participated in the study design, and data gathering, analysis, and interpretation. All authors had full access to the data and are responsible for the veracity and completeness of the reported data. The corresponding author had final responsibility for the decision to submit for publication.
Results
Of 324 patients who were screened, 244 antiretroviral-therapy-naive patients were randomly allocated to one of the four treatment regimens; 243 patients received at least one dose of study drug and were included in the analysis (intention-to-treat exposed population; figure 1). Baseline demographics and disease characteristics were balanced across treatment groups (table 1). The maintenance phase had 207 patients (intention-to-treat maintenance-exposed population) and no meaningful differences were noted in the baseline characteristics compared with the overall population (appendix). By the end of the induction phase (week 24), 15 (24%) of 62 patients in the efavirenz group and 21 (12%) of 181 in the cabotegravir groups discontinued treatment (figure 1). During 96 weeks, 21 (34%) of 62 patients in the efavirenz group and 35 (19%) of 181 patients in the cabotegravir groups withdrew from the study. The higher withdrawal rate in the efavirenz group was mainly because of more adverse events (nine [15%]) than in the cabotegravir groups (six [3%]), including four adverse events in the 60 mg cabotegravir group (figure 1).
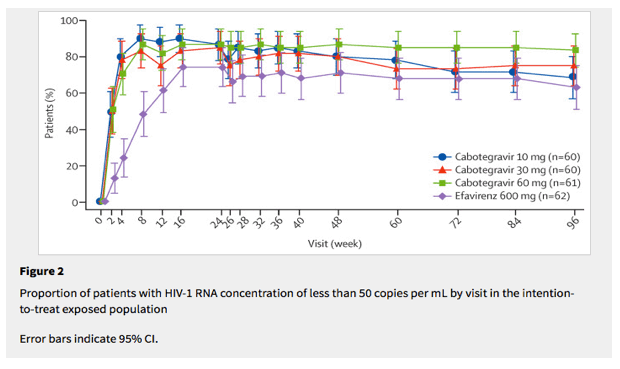
The viral response was robust (HIV-1 RNA <50 copies per mL) in all cabotegravir plus NRTI groups by the end of the 24-week induction phase (intention-to-treat exposed population: 156 [86%] of 181 patients in the cabotegravir groups vs 46 [74%] of 62 patients in the efavirenz group; figure 2; appendix), with a shorter time to viral suppression in the cabotegravir groups than in the efavirenz group (log-rank p<0·0001; figure 2). The advantage in viral response for patients treated with cabotegravir was sustained for an additional 72 weeks after discontinuation of their background NRTIs and switch to the two-drug maintenance regimen (cabotegravir plus rilpivirine; appendix).
For the primary efficacy endpoint, the proportion of patients with plasma HIV-1 RNA of fewer than 50 copies per mL (FDA snapshot algorithm) in each of the cabotegravir plus rilpivirine groups remained numerically higher than in the efavirenz plus dual NRTI group after 24 weeks of induction and 24 weeks of maintenance therapy (week 48; 149 [82%] of 181 patients in the cabotegravir group vs 44 [71%] of 62 patients in the efavirenz group; appendix). After 72 weeks of the two-drug maintenance therapy (week 96), 137 (76%) of patients given cabotegravir plus rilpivirine and 39 (63%) of those continued on efavirenz plus dual NRTI remained virologically suppressed (table 2). Differences in responses until the end of week 96 resulted mainly from an excess of adverse-event-related discontinuations in the efavirenz group (six [3%] in cabotegravir groups vs eight [13%] in efavirenz group), and a lower rate of virological non-responders in the cabotegravir group (18 [10%] vs ten [16%] in the efavirenz group; table 2). At week 96, 51 (84%) of 61, 45 (75%) of 60, and 41 (68%) of 60 patients in the cabotegravir 60 mg, 30 mg, and 10 mg groups, respectively, had a viral response (table 2). Differences in response rates between dose groups were mainly due to rates of viral non-response and study discontinuations for other reasons in the absence of viral data (table 2). Other reasons were protocol deviation, loss to follow-up, investigator discretion, lack of efficacy (viral load at the time of withdrawal was <50 copies per mL), and withdrawal of consent.
For patients with HIV-1 RNA concentrations of fewer than 100 000 copies per mL at baseline, of those receiving cabotegravir, 43 (88%) of 49 in the 60 mg group, 40 (75%) of 53 in the 30 mg group, and 37 (71%) of 52 in the 10 mg group had sustained viral suppression after 72 weeks of maintenance therapy (week 96), compared with 32 (59%) of 54 patients receiving efavirenz. For patients who had a high viral load (HIV-1 RNA of at least 100 000 copies per mL) at baseline, of those receiving cabotegravir, eight (67%) of 12 in the 60 mg group, five (71%) of seven in the 30 mg group, and four (50%) of eight in the 10 mg group had sustained viral suppression at 72 weeks, compared with seven (88%) of eight patients in the efavirenz group. Patients in the cabotegravir groups with a high viral load were discontinued for both viral and non-viral reasons. Two of the patients discontinued had viral loads of greater than 2 million copies per mL at baseline and were not eligible to enter the maintenance phase at week 24 because of insufficient virological response. One of these patients (in the 60 mg cabotegravir group) had 2 158 103 copies per mL of HIV-1 RNA at baseline, 108 copies per mL at week 20, and 59 copies per mL at an unscheduled retest at week 22. The other patient (in the 10 mg cabotegravir group) with 8 425 886 copies per mL of HIV-1 RNA at baseline had 189 copies per mL at week 20, and 329 copies per mL at a week 20 retest. Two patients (in the 60 mg and 10 mg cabotegravir groups) had viral loads of 50-100 copies per mL at week 96 and one patient (in the 10 mg cabotegravir group) had missing viral load data at week 96. Five additional patients in the cabotegravir groups with a high viral load at baseline were discontinued for non-viral reasons (three relocations, one adverse event, and one protocol deviation). The patient with the high viral load allocated to efavirenz had a reduction in HIV-1 RNA from 341 839 copies per mL at baseline to 58 copies per mL at week 26 (withdrawal visit). This patient was discontinued by the investigator at week 26 because of a slow viral response.
For patients treated with a background of NRTIs abacavir-lamivudine during induction, 54 (79%) of 68 in the cabotegravir groups and 13 (57%) of 23 in the efavirenz group remained virologically suppressed at week 96. For patients initiating the study with tenofovir-emtricitabine, 83 (73%) of 113 in the cabotegravir groups and 26 (67%) of 39 in the efavirenz group remained virologically suppressed at week 96.
An efficacy analysis of the intention-to-treat maintenance exposed population at week 96 to assess the two-drug regimen for the maintenance of viral suppression showed similar viral responses between the treatments (137 [86%] of 160 patients in the cabotegravir groups and 39 [83%] of 47 patients in the efavirenz group; table 3) with numerically higher values for the cabotegravir 30 mg (45 [85%] of 53 patients) and 60 mg (51 [93%] of 55 patients) groups compared with the 10 mg group (41 [79%] of 52 patients).
After 24 weeks of induction therapy, the median increase in CD4 cell count from baseline was 185·0 per μL (IQR 95·0-270·0) in the cabotegravir groups and 159·0 cells per μL (43·0-212·0) in the efavirenz group. After 24 weeks of maintenance therapy (week 48), median increase in CD4 cell count from baseline was 219·0 per μL (141·0-343·0) for patients given cabotegravir plus rilpivirine and 216·0 cells per μL (133·5-363·0) for those given efavirenz plus dual NRTIs. By week 96, median increase in CD4 cell count from baseline was 259·5 cells per μL (137·0-355·0) for patients given cabotegravir plus rilpivirine and 289·0 cells per μL (158·0-415·0) for those given efavirenz plus NRTIs. The appendix provides CD4 cell counts at baseline and at weeks 24, 48, 72, and 96.
C0 in the cabotegravir group increased proportionally with the dose. The geometric mean C0 was eight, 24, and 50 times higher than the in-vitro protein-adjusted 90% inhibitory concentration (PA-IC90) of 0·166 μg/mL for cabotegravir 10 mg, 30 mg, and 60 mg, respectively, during maintenance (week 36), with bioequivalent exposures during induction (appendix). Rilpivirine geometric mean individual average C0 at week 36 was five to seven times higher than in-vitro PA-IC90 of 12 ng/mL after administration of cabotegravir 10 mg, 30 mg, and 60 mg (appendix).
During induction, seven patients met the criteria for protocol-defined virological failures: one patient in each of the cabotegravir dose groups and four patients in the efavirenz group. Genotypic or phenotypic resistance did not emerge in any of these patients. Five patients met criteria for protocol-defined virological failures during maintenance: two patients in the cabotegravir 10 mg group (weeks 48 and 72), one patient in the 30 mg group (week 36), and two patients in the efavirenz group (weeks 36 and 60). Treatment-emergent NNRTI (E138Q) and INI (Q148R) resistance mutations were noted in one patient with protocol-defined virological failures at week 48 who was receiving 10 mg cabotegravir. The patient had a 2·04 fold change in sensitivity to etravirine, a 1·83 fold change in sensitivity to rilpivirine, a 3·08 fold change in sensitivity to cabotegravir, and a 30 fold change in sensitivity to raltegravir at week 48. This first patient had cabotegravir and rilpivirine exposures of less than 50% of the study average during the induction and maintenance phases. The second patient who was receiving cabotegravir 10 mg plus rilpivirine met protocol-defined virological failure at week 72 and had treatment-emergent NNRTI-resistance mutations K101K/E and E138E/A with an accompanying 4·6 fold change in sensitivity to rilpivirine. A third patient who was receiving cabotegravir 10 mg plus rilpivirine was lost to follow-up before confirmation of virological failure. Genotype and phenotype analyses on week 48 samples from this patient showed treatment-emergent NNRTI-resistance mutations K101K/E and E138E/K and accompanying rilpivirine phenotypic resistance (2·18 fold change in sensitivity); however, available samples were not sufficient to generate INI-resistance profiles. Neither patient on efavirenz nor the patient on cabotegravir 30 mg plus rilpivirine had treatment-emergent resistance mutations. Six patients were enrolled in this study despite having the primary rilpivirine resistance mutation E138A at screening. All six patients had viral suppression to less than 50 copies per mL HIV-1 RNA at week 96, including four on cabotegravir (two in the 10 mg group and two in the 60 mg group) and two on efavirenz.
The most common treatment-emergent clinical adverse events reported during both the induction and maintenance phases are listed in table 4. Treatment-related adverse events of any grade until the end of week 96 were reported by more patients receiving efavirenz plus NRTIs (42 [68%] of 62) than by those receiving cabotegravir with NRTIs followed by rilpivirine (93 [51%] of 181; table 4).
Treatment-related adverse events reported at an incidence of 10% or greater included headache, nausea, and diarrhoea for cabotegravir and dizziness, abnormal dreams, nausea, fatigue, and insomnia for efavirenz (table 4). Headache was reported in 40 (22%) of 181 patients in the cabotegravir groups compared with seven (11%) of 62 patients in the efavirenz group. Most of the headaches in the cabotegravir groups were transient and mild (grade 1: 29 [16%] of 181) to moderate (grade 2: nine [5%] of 181) with two (1%) grade 3 headaches (in 30 mg and 60 mg groups) compared with no grade 3 headaches in the efavirenz group. The incidences of headaches were similar between the cabotegravir dose groups. Cases of nausea in the cabotegravir groups were mild to moderate with 33 (18%) patients reporting grade 1 events and nine (5%) reporting grade 2 events. 33 (18%) patients in the cabotegravir groups reported grade 1 diarrhoea, nine (5%) reported grade 2 events, and one (1%) patient in the 10 mg group reported grade 3 diarrhoea. Rates of depression were similar (6-10%) across all treatment groups. With the possible exception of insomnia, no association was noted between cabotegravir doses and frequencies of individual adverse events. For patients in the cabotegravir groups, treatment-related adverse events decreased from 74 (46%) of 160 during the 24-week induction phase to 29 (18%) of 160 during the maintenance phase (appendix). Treatment-related adverse events also decreased for patients in the efavirenz group from 32 (68%) of 47 during the induction phase to five (11%) during the maintenance phase. The only treatment-related adverse events reported with a frequency of at least 5% were nausea (four [7%] of 55) and abnormal dreams (three [5%]) in the cabotegravir 60 mg group during the maintenance phase. Serious adverse events occurred in 19 (10%) of 181 cabotegravir-treated patients (none drug related) and in four (6%) of 62 efavirenz-treated patients (one treatment-related suicide attempt; appendix). The only serious adverse events occurring in more than one patient in any treatment group was cellulitis (two [3%] patients in cabotegravir 10 mg group).
Treatment-emergent laboratory abnormalities of at least grade 3 occurred in 47 (26%) of cabotegravir-treated and 23 (37%) of efavirenz-treated patients until week 96 (appendix). Grade 1 or 2 treatment-emergent alanine aminotransferase abnormalities were more common in the cabotegravir 60 mg group (15 [25%] of 61 patients) than in the 30 mg (11 [18%] of 60 patients) or 10 mg groups (eight [13%] of 60 patients) or in the efavirenz group (12 [19%] of 62 patients). Two patients receiving cabotegravir 60 mg plus abacavir-lamivudine met protocol-defined stopping criteria for liver disease of alanine aminotransferase greater than eight times the upper limit of normal (week 4 and week 8). Both patients had grade 1 alanine aminotransferase values at baseline and fatty liver diagnosed with imaging (one before and one after study entry), which acted as potential confounders. Neither of these patients had hepatitis co-infection at baseline nor during treatment. Another patient receiving cabotegravir 30 mg met stopping criteria at week 96. This patient was diagnosed with acute hepatitis C infection, which was judged to be not related to study drug. One patient receiving efavirenz had increased liver transaminases at the week 84 visit and on retest, and met stopping criteria with grade 4 increases for both alanine aminotransferase and aspartate aminotransferase. This patient had concurrent hepatitis C and syphilis infections, and the increase in liver chemistry was not thought to be drug related. Grade 1 to 2 increases in total bilirubin were reported in eight (13%) of 61 patients in the cabotegravir 60 mg group, three (5%) of 60 patients in the 30 mg group, and eight (13%) of 60 patients in the 10 mg group, compared with no increases reported for patients treated with efavirenz. No pattern of incidence or grade of alanine aminotransferase abnormalities was apparent by cabotegravir dose.
Discussion
At week 24 of the induction phase of LATTE, virological response rates were higher in the cabotegravir group than in the efavirenz (control) group because of fewer viral non-responders and adverse-event-related withdrawals in the cabotegravir group. All treatment groups had viral suppression until the end of 72 weeks of maintenance therapy (week 96), with overall suppression rates remaining numerically higher for cabotegravir groups. Response rates at week 96 increased with cabotegravir doses, driven by differences in viral response and non-viral discontinuations between cabotegravir groups. The results of the phase 2b SPRING-123 and phase 3 SINGLE24 studies showed higher viral response rates (time to loss of viral response algorithm) in antiretroviral-therapy-naive patients treated with dolutegravir than in those treated with efavirenz plus dual NRTI control. Together, results from SPRING-1, SINGLE, and LATTE provide a consistent profile of long-term viral response with treatment regimens, including the INI analogues of dolutegravir and cabotegravir compared with efavirenz.
Results from LATTE indicating that the two-drug regimen of cabotegravir plus rilpivirine provides viral suppression that is at least similar to the three-drug regimen of efavirenz plus dual NRTIs for 72 weeks of maintenance therapy in an antiretroviral-naive adult population are informative for further assessment and development of longacting injectable formulations of cabotegravir and rilpivirine. Subgroup analyses by baseline NRTI backbone were consistent with the overall treatment differences between groups. Although differences were noted in the efficacy of cabotegravir and efavirenz between patients with a high viral load (≥100 000 copies per mL) versus low viral load (<100 000 copies per mL) at baseline, subgroup numbers for these strata were small and should be interpreted with caution.
One patient in the cabotegravir 10 mg group developed treatment-emergent resistance mutations Q148R and E138Q at week 48 coinciding with low plasma cabotegravir and rilpivirine throughout both the induction and maintenance phases. Additionally, immediately before virological failure, the patient began a severe, calorie-restricted diet between weeks 40 and 48 that could be predicted to further reduce the concentrations of rilpivirine, if not taken with food. Whether non-compliance played a part in the lower than expected drug concentrations and loss of viral suppression is not known. Q148R has not been detected in any other patients receiving cabotegravir so far; hence, we cannot explore the implications of very low cabotegravir and rilpivirine plasma concentrations selecting for this mutation. Although structurally similar to dolutegravir, results from in-vitro studies with a site-directed mutant showed that cabotegravir has a higher fold change and reduced dissociative half-life with Q148R compared with dolutegravir. Conformational flexibility of the metal-chelating scaffold of dolutegravir versus the more rigid scaffold of cabotegravir might contribute to this difference.25 All three patients with confirmed INI or NNRTI resistance mutations were in the cabotegravir 10 mg group.
Cabotegravir was generally well tolerated with few adverse-event-related withdrawals or discernible trends of adverse events relative to dose. Reported rates of insomnia suggested an association with cabotegravir doses although the rate of insomnia for the highest dose of cabotegravir (60 mg) was lower than the rate of insomnia reported in the efavirenz group (table 4); insomnia is a common adverse event with efavirenz.26 Mild to moderate headaches (grade 1 or 2) were reported more frequently in the cabotegravir group than in the efavirenz group. Nervous system symptoms including rates of insomnia and headache will be assessed closely in future cabotegravir trials. Three patients treated with cabotegravir met protocol-defined liver stopping criteria. The patient receiving 30 mg was diagnosed with acute hepatitis C infection, which was not thought to be treatment related. The two patients receiving cabotegravir 60 mg had underlying steatohepatitis; however, treatment-relatedness of their increases in alanine aminotransferase concentrations (more than eight times the upper limit of normal) could not be ruled out, and increases in alanine aminotransferase resolved on withdrawal of the study drug. Although overall rates of grade 3 and 4 increases in alanine aminotransferase were similar between the cabotegravir and efavirenz groups, transaminases will be closely monitored in ongoing and future studies with cabotegravir. Mild (grade 1 to 2), non-progressive bilirubin increases in a small subset of patients might be a consequence of cabotegravir acting as a substrate for UDP glucoronosyltransferase 1 family polypeptide A1 (UGT1A1 with some involvement from UGT1A9). Cabotegravir, an inhibitor of UGT1A3 and UGT1A9 with 50% inhibitory concentrations of 12 μmol/L and 46 μmol/L, respectively, has also showed some weak inhibition of UGT1A1 and UGT2B17. As a substrate of UGT1A1, bilirubin concentrations might be affected by cabotegravir, but the effect is expected to be small. In LATTE, grade 1 and 2 increases in bilirubin were not dose dependent, with similar incidences across doses of 10-60 mg.
The substitution of dual NRTIs by rilpivirine in the cabotegravir groups at week 24 did not lead to an increase in the rate of adverse events or laboratory abnormalities. A comparison of drug-related adverse events between the induction and maintenance phases indicated that cabotegravir plus rilpivirine and efavirenz plus dual NRTIs were well tolerated throughout the maintenance phase.
Although patients receiving cabotegravir and their investigators were masked to the dose of cabotegravir, a limitation of the LATTE study design was that neither patients nor investigators were masked to whether the participants received cabotegravir or efavirenz, potentially affecting rates of patient retention and reporting of adverse events. The incidence of adverse events reported for efavirenz within the LATTE study was generally consistent with the established safety profile for efavirenz.26
On the basis that efficacy, safety and tolerability, viral resistance, and pharmacokinetic measures were similar between the three cabotegravir dose groups, an a priori criteria determined that the 30 mg once a day dose would be selected for further assessment. Although response rates were numerically different for 10 mg, 30 mg, and 60 mg between weeks 48 and 96, by review of the Snapshot results (table 2), a detailed breakdown of the response rates showed that much of the differences between dosing groups were due to a higher rate of discontinuations for non-viral reasons in the 10 mg and 30 mg groups. The rate of viral non-response was higher in the cabotegravir 10 mg group (15%) than in the 30 mg (10%) and 60 mg groups (5%; table 2). Three patients receiving cabotegravir 30 mg were counted as Snapshot non-responders because of a change in background antiretroviral therapy (table 2), two of whom remained virally suppressed (HIV-1 RNA <50 copies per mL) for 96 weeks. Counting these patients as non-responders contributed to a slightly reduced virological success rate for the cabotegravir 30 mg group. Additionally, differences in response were noted in the no viral data category for patients discontinuing for other reasons (including protocol deviations, lost to follow-up, and withdrawal of consent): cabotegravir 10 mg (13%) and 30 mg (13%) compared with cabotegravir 60 mg (5%; table 2). More patients in the cabotegravir 60 mg group withdrew early from the study because of adverse events (2% in each of the 10 mg and 30 mg groups and 7% in the 60 mg group). Although viral suppression was longlasting with all cabotegravir doses, the combined efficacy and safety results until the end of week 96 lend support to the selection of oral cabotegravir 30 mg once a day for further assessment. All patients still receiving cabotegravir were changed over after week 96 to cabotegravir 30 mg plus rilpivirine in the ongoing open-label phase of LATTE.
Cabotegravir 30 mg once a day orally, cabotegravir longacting, and rilpivirine longacting formulations are currently being investigated in the LATTE-2 clinical trial with the goal of extending the LATTE results and confirming an appropriate oral lead-in and longacting dosing regimen for assessment in the planned phase 3 programme. Results from LATTE have formed the foundation for the first assessment of a two-drug all-injectable HIV-1 regimen with possibilities for improved therapeutic intervention.
Contributors
All authors contributed to data interpretation, drafting the manuscript, and revising it for critical content.
Declaration of interests
DAM, SKG, MHSC, KJH, SLF, BSS, MMB, and WRS were employees of GlaxoSmithKline at the time the study was being done and the manuscript was being written. DAM, SKG, MHSC, KJH, and WRS are employed by ViiV Healthcare. SLF, BSS, and MMB are employed by PAREXEL International. MMB received grants, personal fees, and non-financial support from GlaxoSmithKline while doing work on the behalf of Janssen and ViiV Healthcare. CCB has served as a clinical trial principal investigator for AstraZeneca, Bristol-Myers Squibb, Gilead Sciences, GlaxoSmithKline, Janssen, Johnson & Johnson, Novartis, Novo Nordisk, Pfizer, Salix, Sangamo, Shionogi, Sliagen, Synergy, Vertex, and ViiV Healthcare, has served on advisory boards for Bristol-Myers Squibb and Gilead Sciences, and has served as an education committee member and speaker for Gilead Sciences. GHRS has received speaker fees from Gilead Sciences. DPH has served on speaker bureaus or advisory boards for AbbVie, Bristol-Myers Squibb, Gilead Sciences, Janssen Therapeutics, Merck, and ViiV Healthcare. JJE has received grants from GlaxoSmithKline and personal fees from Bristol-Myers Squibb, Gilead Sciences, GlaxoSmithKline, Janssen, Merck, and ViiV Healthcare. MCS is an employee of Janssen Infectious Diseases and owns stock and stock options in Johnson & Johnson. PEW is an employee of and owns stock in Johnson & Johnson. KYS is an employee of ViiV Healthcare. JdV declares no competing interests.
|
|
| |
| |
|
|
|