 |
|
|
| |
The 17th International Workshop on Clinical Pharmacology of HIV and Hepatitis Therapy Report by Courtney Fletcher, PharmD/Jennifer Kiser, PharmD
|
| |
| |
Courtney V. Fletcher, Pharm.D.
Dean and Professor
College of Pharmacy
University of Nebraska Medical Center
Jennifer J. Kiser, Pharm.D.
Associate Professor
School of Pharmacy
University of Colorado at Denver
The 17th International Workshop on Clinical Pharmacology of HIV & Hepatitis Therapy was held in Washington, DC, from June 8-10, 2016. In this report we will highlight abstracts focused on pharmacologic issues that are of broad interest or might benefit from some expert clarification. Abstracts will be discussed in two general categories: (i) the clinical pharmacology of therapy for HIV infection, and (ii) the clinical pharmacology of therapy for hepatitis infection. This report does not cover all plenaries and data presented at the Workshop. For more information and to view presentations from the meeting please visit:
http://www.infectiousdiseasesonline.com/17th-hivhep-pk-presentations/
The Clinical Pharmacology of Therapy for HIV Infection.
Courtney V. Fletcher, Pharm.D.
A. DRUG DEVELOPMENT
1. Revised dosing for cabotegravir-long acting (CAB-LA) and rilpivirine-long acting (RPV-LA) for maintenance of HIV suppression.
At this meeting, new information was presented by Bill Spreen, Pharm.D. with ViiV Health Care and Herta Crauwels, Ph.D. with Janssen on dosing interval selection and revised doses for both CAB-LA and RPV-LA. For the upcoming Phase III trials for maintenance of HIV suppression, an every four-week interval has been selected for both CAB-LA and RPV-LA, and the loading doses for both will be increased. The every eight-week dosing interval has not been abandoned, and will be evaluated in future studies.
The LATTE-2 study for maintenance of HIV-suppression with IM formations of CAB-LA and RPV-LA were presented at CROI 2016 (Abstract 31LB). Briefly, 309 HIV-infected individuals were randomized to receive CAB-LA and RPV-LA either every 4-weeks or every 8-weeks, or to remain on the oral induction regimen. All participants had HIV-RNA < 50 copies/mL with an induction regimen of oral CAB (30 mg once daily) given with ABC+3TC. The every 4-week CAB-LA and RPV-LA doses were 400 mg and 600 mg, respectively; the every 8-week doses were 600 mg and 900 mg, respectively. The CAB-LA formulation is 200 mg/mL and the RPV-LA is 300 mg/mL; injection volumes, therefore, for the every 8-week regimen were 3 mLs for CAB-LA and 3mLs for RPV-LA. At week 32, the proportions of participants with HIV-RNA < 50 copies/mL were: oral CAB/ABC/3TC, 91%; every 4-week CAB-LA and RPV-LA, 94%; and every 8-week CAB-LA and RPV-LA, 95%. A clear upward trend over time in RPV plasma concentrations with LA administration was noted; concentrations during the first 16 weeks were lower than those from 16-32 weeks. Though not as pronounced, an upward trend over time was apparent for CAB plasma concentrations as well.
Increased Loading Doses of CAB-LA and RPV-LA. For both CAB-LA and RPV-LA, a larger loading dose will now be given on day 1, followed by the same every four-week maintenance doses used in the LATTE-2 study. CAB-LA: the dose for the upcoming Phase III trials will be a loading dose of 600 mg IM, followed by 400 mg IM every four weeks. RPV-LA: the dose of RPB-LA will be a loading dose of 900 mg IM, followed by 600 mg IM every four weeks. These revised dosing regimens will be used in two Phase III trials, to be called FLAIR and ATLAS, which are switch studies from an oral ARV regimen to the CAB-LA + RPV-LA regimen. These studies are planned to start in the 3rd quarter of 2016.
CROI: ECLAIR: Phase 2A Safety and PK Study of Cabotegravir LA in HIV-Uninfected Men.......Long-Acting Cabotegravir Data Suggest PrEP Injections Every 8 Week - Mark Mascolini - (02/24/16)
CROI: Cabotegravir + Rilpivirine as Long-Acting Maintenance Therapy: LATTE-2 Week 32 Results - (02/24/16)
Pharmacology Wk: Cabotegravir Long-Acting (LA) Injectable Nanosuspension - (06/16/15)
2. Macaque pharmacokinetic and virologic data support weekly oral dosing of MK-8591.
MK-8591 (or EFdA) is a NRTI with very potent in vitro activity against HIV: the IC50 is approximately 0.2 nM, which would make it approximately 8400 times more potent than tenofovir (see Antimicrob Agents Chemother 2015;59:4190. doi: 10.1128/AAC.05036-14). In macaques, preliminary PK studies indicated an estimated plasma half-life of 7 hours, and an intracellular half-life of EFdA-triphosphate (EFdA-TP) of > 72 hours. MK-8591 is presently being developed by Merck.
Abstract #O_13 evaluated the virologic activity of MK-8591 in macaques given once weekly oral doses ranging from 1.3 to 18.2 mg/kg, or a once daily oral dose of 0.19 mg/kg. The maximal virologic decline (approximately 1.5 log decrease in viral load) was seen with weekly doses of 3.9 to 18.2 mg/kg once weekly, and was correlated with intracellular-triphosphate concentrations of MK-8591 of ≥ 0.53 pmol/10 million cells. Viral suppression was maintained for at least 7 days after the last dose. This study provides proof of concept for a once weekly oral dosing strategy of MK-8591. In Q&A, the authors stated their plan to develop MK-8591 for prevention and treatment, and acknowledged for the latter they would need other agents to form a complete, once weekly regimen and that they were committed to that goal. I think MK-8591 is a promising agent and is one to keep your eye on.
PharmacologyWk: Efficacy of once-weekly MK-8591 in SIV infected rhesus macaques - (06/14/15)
B. HIV Pharmacotherapy in special populations
1. DRV/RTV dosing in pregnant women: 600mg/100mg twice daily is recommended.
Dr. Minh Le reported on the PK, safety and efficacy of DRV/RTV given to 220 HIV-infected pregnant women, 89% from sub-Saharan Africa (Abstract O_1). This work is significant because it represents the largest study of DRV/RTV in pregnant women to my knowledge. During pregnancy, women remained on DRV/RTV 600 twice daily (n=71); on 800/100 once daily (n=88), or switched from 800/100 once daily to 600/100 twice daily (n=61). For women who remained on twice daily or switched to twice daily, DRV trough concentrations remained consistent across trimesters and delivery (≈ 2000 ng/mL). However, for those who remained on once daily, there was a steady decline during pregnancy to delivery in trough concentrations (median 1st trimester, 1574 ng/mL; 2nd trimester, 1144 ng/mL; 3rd trimester, 934 ng/mL; and at delivery, 854 ng/mL). Overall, 96% of women had HIV-RNA < 400 copies/mL at delivery and no mother-to-child HIV transmission has been found to date.
DRV/RTV is a preferred PI for use in pregnant women, and DRV/RTV concentrations are reduced during pregnancy compared with postpartum. The DRV/RTV 600/100 twice daily regimen compared with 800/100 once daily, achieves higher trough concentrations and appears to maintain more equivalent concentrations across trimesters than does the once daily. Notably, from the study of Le at this meeting, only 9% of women receiving twice daily DRV/RTV had trough concentrations at delivery less than 550 ng/mL, the suggested threshold concentration, versus 30% of those receiving once daily. The DHHS Perinatal Guidelines presently recommend using the DRV/RTV 600mg/100mg twice-daily regimen in pregnant women; they do not recommend DRV/RTV once daily. On June 17, 2016, the FDA revised their guidelines for DRV in pregnant women and also recommends the 600/100 twice-daily regimen (see: http://www.accessdata.fda.gov/drugsatfda_docs/label/2016/021976s043,202895s017lbledt.pdf ). They, however, state that 800/100 once daily could be considered in women already stable on the once daily regimen and in whom a change to twice daily would compromise tolerability or compliance.
I agree with the recommendations of the FDA and the DHHS that DRV/RTV 600/100 twice daily is the recommended regimen. While no treatment failures were reported in the study Dr. Le presented at this conference with the 800/100 once daily regimen, the lower troughs achieved and the increased proportion with concentrations less than a suggested threshold, create some increased risk of failure. I see no reason to pose that increased risk of maternal-to-child transmission, except in rare/very select circumstances.
C. DRUG-DRUG INTERACTIONS
1. Switching from an EFV-containing regimen to a DTG-containing one? No dose adjustment of DTG is necessary, but come cautionary comments.
The dose of DTG should be increased from 50 mg once daily to 50 mg twice daily if given with EFV in order to compensate for the ≈ 60% and 75% decrease in AUC and trough DTG concentrations, respectively. Abstract O_23 investigated whether a dose increase of DTG was necessary in persons who were switching from an EFV-containing regimen to a DTG-containing one. 24 HIV-infected adults on an EFV-containing regimen were switched to ABC/3TC/DTG, and had virologic and PK evaluations for 24 weeks after the change. All participants had HIV-RNA < 50 copies/mL at baseline and all remained < 50 copies/mL at each time point evaluated over the 24-week post-switch study period. Mean baseline EFV concentrations were 1830 ng/mL, and declined to 320 ng/mL at week 1, and were undetectable in all subjects at week 8. After the switch, mean (and SD) DTG trough concentrations at 1 week were 938 (± 955) ng/mL and by week 4 rose to steady-state values sustained between 1186-1378 (± 710 to 1018) ng/ml from week 4 to 24.
In general, these virologic and PK data support some virologically-suppressed individuals on an EFV-containing regimen can be switched to a DTG-containing regimen without the need for a short-term increase in the DTG dose while EFV concentrations decay. But I have a few cautionary comments. First, this was a small study was done in the US, Canada and Puerto Rico. Based on the mean (SD) EFV concentration at baseline of 1830 (±660) ng/mL, it does not appear individuals with the slow metabolizing CYP2B6 polymorphism more common among black persons were represented. For example, in a study in adults and children in South Africa, median EFV concentrations were 1440, 2080 and 7260 ng/mL for the extensive, intermediate and slow metabolizers, respectively. (See Sinaxdi PZ, Br J Clin Pharmacol, July 2015, http://www.ncbi.nlm.nih.gov/pubmed/25611810).
Individuals with the slow metabolizer genotype will have higher EFV concentrations at baseline, a slower decay of EFV after discontinuation, and may have a more prolonged period of lower DTG concentrations and a slower rise to DTG steady-state concentrations. Similar virologic and PK data on switching from EFV to DTG in a larger and more multi-ethic represented population are needed. Second, it seems to me there is greater intersubject variability in DTG concentrations that what is usually reported. Take a look at slide #8 from the presentation (see http://regist2.virology-education.com/2016/17HIVHEPPK/39_deWet.pdf) and you can see certain individuals who had DTG trough concentrations equal to or perhaps slightly less than the IC90 of 64 ng/mL, at week 1 while EFV was still present, but also at week 8. While the virologic data provide evidence of suppression, the PK data raise some questions about prolonged metabolism induction effects of EFV and/or greater DTG interpatient PK variability. For clinicians, if there is any concern about virologic suppression (previous blips), other concomitant medications that might lower DTG concentrations (Mg or Al antacids, or oral Ca or iron supplements), a short course (one to two weeks) of 50 mg twice daily DTG could be a prudent safeguard to help ensure the best possible virologic outcome for a person switching from EFV to DTG.
2. Rifampin is not recommended for administration with oral CAB or CAB-LA.
Rifampin (RIF) is a potent inducer of drug metabolizing enzymes, decreasing the concentrations of many ARVs such that co-administration is not possible. A PK study of RIF given with oral CAB was conducted to evaluate the effects of RIF on CAB plasma concentrations (Abstract O_18). 15 healthy volunteers received a single 30 mg dose of oral CAB alone; this dose was repeated with RIF following 12 days of RIF administration at 600 mg once daily. CAB concentrations were significantly decreased in the presence of RIF: the ratio (with RIF to without RIF) of the AUC for CAB was 0.41, indicating CAB plasma concentrations were reduced 59% (more than half). This magnitude of a reduction is similar to that seen when RIF is given with RAL and DTG, resulting in 40% and 54% reductions in concentrations, respectively. The elimination half-life of CAB was reduced from 39 hours alone to 16 hours with RIF coadministration. These data provide clear evidence that RIF potently induces the metabolism of CAB and should not be administered with oral CAB. This drug-drug interaction study was intended also to inform on the nature of the interaction between RIF and CAB-LA. Such extrapolation is necessary because the very long half-life of the LA formulation (20-40 days) would require such a lengthy time for participant participation (6+ months) as to likely be unworkable for recruitment/retention. These data provide high confidence that a significant reduction in CAB plasma concentrations would be expected with the combination of RIF with CAB-LA, and this combination is not recommended.
PharmacologyWk: Rifampin (RIF) Decreases Cabotegravir (CAB) Exposure following Oral Coadministration - (06/17/15)
The Clinical Pharmacology of Therapy for Hepatitis.
Jennifer J. Kiser, PharmD
A. HCV PHARMACOTHERAPY IN SPECIAL POPULATIONS
1. PK Studies of DAAs in Renal Impairment Should Not Be Limited to Those on Hemodialysis
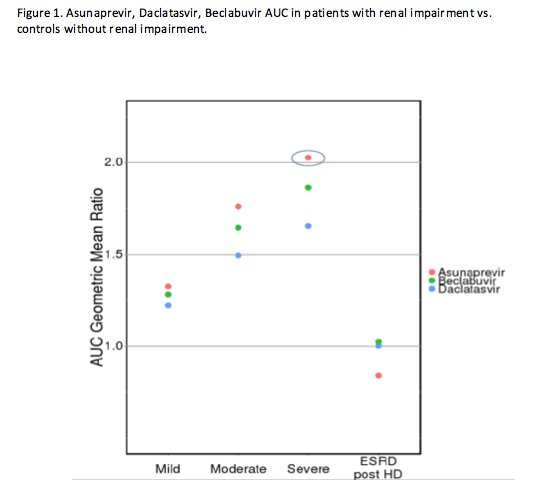
The majority of commercially available direct-acting antiviral agents (DAAs) are metabolized by the liver. With the exception of sofosbuvir, less than 10% of a dose of other DAAs is eliminated by the kidneys. Despite this, the exposures of most DAAs are increased in individuals with renal impairment. The exact mechanism for this is unclear, but the accumulation of uremic toxins, parathyroid hormone, and/or cytokines in those with renal impairment may impair hepatic enzyme or transporter function. Whether the increase in exposures poses a safety risk will depend on the drug, but Dr. Garimella (Abstract #O_05) demonstrated using asunaprevir, daclatasvir, and beclabuvir that limiting the design of renal impairment studies to those with end stage renal disease on hemodialysis (ESRD + HD) misses the true effect of renal impairment on the PK of these DAAs. Figure 1 shows the ratio of the exposures of DAAs in those with renal impairment vs. volunteers with no impairment. On the far right the ratio is close to 1, indicating little change in exposures in those with ESRD+HD relative to volunteers with no impairment. This is likely because HD actually clears the compounds that impair hepatic metabolism and/or transporter function. Had the study not included those with mild (eGFR 60-89 mL/min/1.73 m2), moderate (eGFR 30-59 mL/min/1.73 m2) and severe (eGFR 15-29 mL/min/1.73 m2) renal impairment not on hemodialysis, we would have falsely concluded that renal impairment had no effect on the PK of these DAAs when in fact, asunaprevir exposures were 2-fold higher in those with severe impairment not receiving HD. Based on these results and the therapeutic range of the drug, it is recommended to reduce the dose of asunaprevir by half in this patient population. Investigators also showed through a literature review that this phenomenon was not unique to DAAs. Other hepatically metabolized drugs have increased exposures in patients with renal impairment as well.
In summary, even for drugs that are primarily hepatically metabolized and undergo minimal renal elimination, PK studies of renal impairment should follow the FDA draft guidance (http://www.fda.gov/downloads/Drugs/.../Guidances/UCM204959.pdf) which recommends either a "reduced" design in individuals with ESRD not receiving HD or a "full" design which includes all levels of renal impairment.
B. DRUG INTERACTIONS
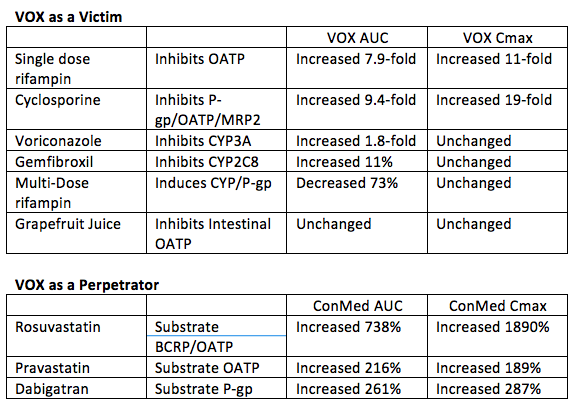
1. DDI with Voxilaprevir (GS-9857)
SOF/Velpatasvir/Voxilaprevir (VOX) is a three-drug fixed dose DAA combination being evaluated in Phase 3 trials. In vitro, VOX is a substrate for CYP3A4, CYP2C8, the OATP uptake transporters and the efflux transporters P-gp and BCRP. VOX is also an inhibitor of P-gp and BCRP in vitro. Dr. Kirby presented drug interaction studies with VOX (Abstracts #O_24 and #O_25). Tables below show results of VEL as a victim and perpetrator in interactions. Results indicate VOX is very sensitive to hepatic inhibition of OATP transporters, so coadministration of VOX with potent OATP inhibitors is not recommended.
Inhibitors of CYP3A4, CYP2C8, and P-gp are likely okay with VOX. As with all HCV treatments, interactions with HMG-CoA reductase inhibitors (statins) must be considered with a potential need to change statins or use a reduced dose of the statin. It is likely based on these data that VOX will have problematic interactions with efavirenz and HIV protease inhibitors.
http://regist2.virology-education.com/2016/17HIVHEPPK/40_Kirby.pdf
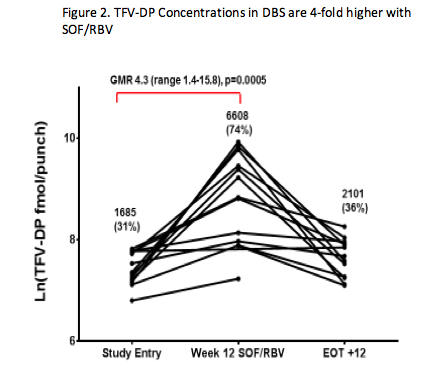
2. SOF Increases Tenofovir Diphosphate Concentrations in Cells
ACTG 5327 (SWIFT-C) is evaluating sofosbuvir (SOF)-containing regimens for the treatment of acute HCV in HIV-infected individuals. In Cohort 1 of SWIFT-C, there was a high rate of relapse to 12 weeks of SOF and ribavirin (RBV) treatment (AASLD 2015 #1094 http://www.natap.org/2015/AASLD/AASLD_108.htm). RBV concentrations were lower in those that relapsed compared to those that achieved SVR (CROI 2016 #99 http://www.natap.org/2016/CROI/croi_192.htm), which given the long plasma half-life of RBV, may indicate lower adherence in those that relapsed. Given the reduced RBV adherence, MacBrayne and colleagues (Abstract #O_19 http://natap.org/2016/Pharm/Pharm_15.htm) sought to evaluate ARV adherence by measuring tenofovir-diphosphate (TFV-DP) concentrations in cells. The long intracellular half-life of TFV-DP gives a measure of long term adherence. Fifteen of 17 patients in Cohort 1 were taking TDF. At study entry, before patients began SOF/RBV treatment, TFV-DP concentrations were consistent with prior studies of individuals taking TDF with good adherence, but after 12 weeks of SOF/RBV treatment, TFV-DP concentrations were 4-fold higher in dried blood spots (Figure 2) and 2-fold higher in PBMCs. TFV-DP concentrations 12 weeks following the completion of SOF/RBV treatment were similar to study entry. Although the intent with this analysis was to estimate TDF adherence, investigators believe this represents an unexpected drug interaction between tenofovir and SOF/RBV. SOF is presumed to be the perpetrator since preliminary data with SOF/ledipasvir (without ribavirin) indicates a similar or greater increase in TFV-DP concentrations. Additional studies are needed to determine the mechanism. It is unclear whether this interaction will have clinical relevance. Creatinine clearance was unchanged from study entry to week 12 of SOF/RBV treatment. These data raise questions about the potential for unrecognized intracellular nucleos(t)ide interactions since drug interaction studies typically measure only plasma concentrations and the plasma concentrations of tenofovir were unchanged in this study. It is also unknown what the implications may be for TAF given it has 20-fold higher TFV-DP concentrations in PBMC.
3. DRV/RTV with PrOD
In healthy volunteers, ritonavir-boosted paritaprevir, ombitasvir, and dasabuvir (PrOD) reduced DRV trough concentrations by 48% with once daily DRV/RTV and 43% with twice daily DRV/RTV. There are conflicting recommendations in the United States prescribing information and the European Union Summary of Product Characteristics (EU SmPC) for this combination based on these data. In the US, this combination is not recommended, whereas in the EU SmPC, DRV/RTV 800/100mg once daily can be given with PrOD in the absence of extensive protease inhibitor resistance. Part Ib of the TURQUOISE-1 study is evaluating this combination in individuals with HIV/HCV coinfection. Twenty-two patients taking once daily DRV/RTV 800/100mg were randomized to stay on once daily vs. switch to DRV/RTV 600/100mg twice daily. Intensive PK sampling was performed in these 22 patients before and during treatment with PrOD. At CROI 2016 (#574 http://www.natap.org/2016/CROI/croi_99.htm), Dr. Wyles showed DRV troughs were 36% and 27% lower with PrOD vs. without in those on once daily and twice daily DRV/RTV, respectively and that there were very few HIV "blips". At this meeting, Dr. King (Abstract #O_20 http://regist2.virology-education.com/2016/17HIVHEPPK/36_King.pdf) presented the actual DRV concentrations in these patients and the relationship of DRV troughs to viral blips. After 4 weeks of PrOD, the geometric mean (%CV) DRV troughs were 1045 ng/mL (63%) and 2590 ng/mL (67%) in those on DRV/RTV once daily and twice daily, respectively. Five patients had at least one HIV RNA value between 40 and 200 copies/mL during PrOD treatment, but no association between DRV concentrations and these blips was observed.
These data are reassuring, especially for treatment naïve patients on twice daily DRV/RTV. However, this PK substudy was small (22 patients) and highly treatment experienced individuals were excluded. To qualify, participants were receiving once daily DRV/RTV so presumably did not have DRV-associated resistance mutations and could have only received one PI in prior ARV regimens. Hopefully, we will have increased confidence in the safe use of this combination for treatment-naïve patients when the parent trial is completed.
4. Dose Adjustments for Concomitant Meds After Completing HCV Treatment
Data presented at this meeting (Abstract #P_42) found that 15% of medications used in persons with HCV had the potential to interact with HCV treatments. Some interactions can be managed by dose adjustment of the concomitant medication. However, once DAA treatment is completed, it is unclear when to re-adjust the dose of the concomitant medication. Dr. Mukherjee presented a novel approach for determining the optimal time to dose escalate amlodipine after completing PrOD treatment (Abstract #O_16). Investigators developed a physiologic-based model that linked drug concentrations to blood pressure measures. They found the effect of ritonavir on amlodipine PK took about 5 days to resolve after discontinuing PrOD, but the average systolic blood pressure was only 3mmHg lower if the amlodipine dose was escalated immediately following discontinuation of PrOD vs. waiting 5 days.
This approach of using PB-PK-PD modeling to study interactions may save significant time and money. Many drug interaction studies are performed in healthy volunteers, but this approach holds great promise for extending our ability to define the magnitude of interactions in a variety of patient populations and clinical scenarios without having to conduct multiple trials in humans.
5. Other Notable DDI Data
⋅ Grazoprevir Levels Increased with TDF/FTC/EVG/COBI - Avoid Use (Abstract #O_22 http://natap.org/2016/Pharm/Pharm_31.htm)
⋅ Daclatasvir Dose with ATV/COBI is 30mg - Same as with ATV/RTV (Abstract #P_43)
⋅ No effect of rifaximin on velpatasvir PK (Abstract #O_08 http://natap.org/2016/Pharm/Pharm_14.htm)
Abbreviations
%CV, percent coefficient of variation
3TC, lamivudine
ABC, abacavir
ACTG, AIDS Clinical Trials Group
ARV, antiretroviral drug
AUC, area under the concentration-time curve
ATV, atazanavir
BCRP, breast cancer resistance protein
CAB, cabotegravir
CAB-LA, long-acting cabotegravir
Cmax, maximum drug concentration
COBI, cobicistat
ConMed, concomitant medication
CYP, cytochrome P450 drug metabolizing enzymes
DAA, direct acting antiviral agent(s)
DDI, drug-drug interaction
DHHS, Department of Health and Human Services
DTG, dolutegravir
DRV, darunavir
eGFR, estimated glomerular filtration rate
EU SmPC, European Union Summary of Product Characteristics
ESRD, end-stage renal disease
EVG, elvitegravir
FDA, Food and Drug Administration
FTC, emtricitabine
GALT, gut-associated lymphatic tissue
HCV, Hepatitis C virus
HD, hemodialysis
IM, intramuscular
MRP2, multidrug resistance protein (efflux transporter)
OATP, organic anion transporting polypeptide (uptake transporters)
PA-IC90, protein-binding adjusted 90% inhibitory concentration
PBMCs, peripheral blood mononuclear cells
PD, pharmacodynamics
P-gp, p-glycoprotein (efflux transporter)
PK, pharmacokinetic
PI, protease inhibitor
PrOD = paritaprevir, ritonavir, ombitasvir, and dasabuvir
RAL, raltegravir
RIF, rifampin
RTV, ritonavir
RBV, ribavirin
RPV, rilpivirine
RPV-LA, long-acting rilpivirine
SOF, sofosbuvir
TAF, tenofovir alafenamide
TDF, tenofovir disoproxil fumarate
TFV-DP, tenofovir diphosphate
Trough, concentration immediately before the next dose
VOX, voxilaprevir (GS-9857)
|
| |
|
|
|
|
|