| |
Sofosbuvir, Velpatasvir, and Voxilaprevir for Previously Treated HCV Infection
|
| |
| |
Download the PDF here
Marc Bourlière, M.D., Stuart C. Gordon, M.D., Steven L. Flamm, M.D., Curtis L. Cooper, M.D., Alnoor Ramji, M.D., Myron Tong, M.D., Natarajan Ravendhran, M.D., John M. Vierling, M.D., Tram T. Tran, M.D., Stephen Pianko, M.D., Meena B. Bansal, M.D., Victor de Lédinghen, M.D., Robert H. Hyland, D.Phil., Luisa M. Stamm, M.D., Ph.D., Hadas Dvory-Sobol, Ph.D., Evguenia Svarovskaia, Ph.D., Jie Zhang, Ph.D., K.C. Huang, Ph.D., G. Mani Subramanian, M.D., Diana M. Brainard, M.D., John G. McHutchison, M.D., Elizabeth C. Verna, M.D., Peter Buggisch, M.D., Charles S. Landis, M.D., Ph.D., Ziad H. Younes, M.D, Michael P. Curry, M.D., Simone I. Strasser, M.D., Eugene R. Schiff, M.D., K. Rajender Reddy, M.D., Michael P. Manns, M.D., Kris V. Kowdley, M.D., and Stefan Zeuzem, M.D., for the POLARIS-1 and POLARIS-4 Investigators*
N Engl J Med June 2017
Background
Patients who are chronically infected with hepatitis C virus (HCV) and who do not have a sustained virologic response after treatment with regimens containing direct-acting antiviral agents (DAAs) have limited retreatment options.
Methods
We conducted two phase 3 trials involving patients who had been previously treated with a DAA-containing regimen. In POLARIS-1, patients with HCV genotype 1 infection who had previously received a regimen containing an NS5A inhibitor were randomly assigned in a 1:1 ratio to receive either the nucleotide polymerase inhibitor sofosbuvir, the NS5A inhibitor velpatasvir, and the protease inhibitor voxilaprevir (150 patients) or matching placebo (150 patients) once daily for 12 weeks. Patients who were infected with HCV of other genotypes (114 patients) were enrolled in the sofosbuvir-velpatasvir-voxilaprevir group. In POLARIS-4, patients with HCV genotype 1, 2, or 3 infection who had previously received a DAA regimen but not an NS5A inhibitor were randomly assigned in a 1:1 ratio to receive sofosbuvir-velpatasvir-voxilaprevir (163 patients) or sofosbuvir-velpatasvir (151 patients) for 12 weeks. An additional 19 patients with HCV genotype 4 infection were enrolled in the sofosbuvir-velpatasvir-voxilaprevir group.
Results
In the three active-treatment groups, 46% of the patients had compensated cirrhosis. In POLARIS-1, the rate of sustained virologic response was 96% with sofosbuvir-velpatasvir-voxilaprevir, as compared with 0% with placebo. In POLARIS-4, the rate of response was 98% with sofosbuvir-velpatasvir-voxilaprevir and 90% with sofosbuvir-velpatasvir. The most common adverse events were headache, fatigue, diarrhea, and nausea. In the active-treatment groups in both trials, the percentage of patients who discontinued treatment owing to adverse events was 1% or lower.
Conclusions
Sofosbuvir-velpatasvir-voxilaprevir taken for 12 weeks provided high rates of sustained virologic response among patients across HCV genotypes in whom treatment with a DAA regimen had previously failed. (Funded by Gilead Sciences; POLARIS-1 and POLARIS-4 ClinicalTrials.gov numbers, NCT02607735 and NCT02639247.)
The majority of patients who are chronically infected with hepatitis C virus (HCV) can now be successfully treated with drugs that directly target viral replication.1,2 Combination regimens of direct-acting antiviral agents (DAAs) provide rates of sustained virologic response exceeding 90%, regardless of HCV genotype, disease stage, or treatment history.3 The proportion of patients who do not have a sustained virologic response to treatment with approved regimens is small, but given the size of the infected population — estimates range up to 150 million people worldwide4 — the absolute number of such patients is substantial and will increase as more patients are treated for HCV infection. There are no approved retreatment options for patients who have previously received a regimen containing an NS5A inhibitor.5 These patients, who represent the majority of patients with recent treatment failures, are of particular concern because the resistance-associated substitutions that are selected by NS5A inhibitors maintain viral fitness long after the end of the failed treatment.
Sofosbuvir is a nucleotide analogue HCV NS5B polymerase inhibitor that, in combination with other DAAs, is approved for the treatment of HCV infection of all genotypes.6-8 Velpatasvir is an HCV NS5A inhibitor with pangenotypic potency.9 The fixed-dose combination of sofosbuvir and velpatasvir provided high rates of sustained virologic response in phase 3 clinical trials and has recently been approved for the treatment of patients with HCV infection of any genotype, with or without cirrhosis, regardless of whether they have received previous treatment with interferon-based therapy.10,11 Voxilaprevir (formerly GS-9857, Gilead Sciences) is a pangenotypic inhibitor of the HCV NS3-NS4A protease.12-14 In phase 2 trials, the combination of sofosbuvir, velpatasvir, and voxilaprevir was effective in a broad range of patients with chronic HCV infection.15-18
We conducted two phase 3 trials to assess the efficacy and safety of the fixed-dose combination of sofosbuvir, velpatasvir, and voxilaprevir for 12 weeks in patients who were chronically infected with HCV of any genotype, including patients with compensated cirrhosis, who have previously received unsuccessful treatment with DAA-based regimens.
Methods
Trial Oversight
The two trials were approved by the institutional review board or independent ethics committee at each participating site and were conducted in compliance with the Declaration of Helsinki, Good Clinical Practice guidelines, and local regulatory requirements. The trials were designed and conducted by the sponsor (Gilead Sciences) in collaboration with the principal investigators, in accordance with the protocols, which are available with the full text of this article at NEJM.org. The sponsor collected the data, monitored trial conduct, and performed the statistical analyses. Independent data and safety monitoring committees reviewed the progress of the trials. All the authors had access to the data and assumed responsibility for the integrity and completeness of the reported data and the fidelity of the trials to the protocols. The initial draft of the manuscript was prepared by a writer employed by Gilead Sciences and by the primary investigators (the first and last authors) with input from all the authors.
Patients
We used identical eligibility criteria for the two trials, with the exception that POLARIS-1 enrolled only patients whose previous treatment included an NS5A inhibitor and POLARIS-4 enrolled patients who had been previously treated with any DAA regimen that did not include an NS5A inhibitor (with the exception that those who had received only a protease inhibitor with peginterferon and ribavirin were not included, since these patients have approved retreatment options). Patients had to have had virologic failure after completing previous treatment of at least 4 weeks’ duration; patients who discontinued owing to adverse events or who had virologic failure because of nonadherence to treatment were not enrolled. All patients provided written informed consent. The full eligibility criteria for both trials are provided in the Supplementary Appendix, available at NEJM.org.
Trial Design
In POLARIS-1, patients were enrolled at 108 sites in the United States, Canada, New Zealand, Australia, France, Germany, and the United Kingdom from November 2015 through May 2016. Patients with HCV genotype 1 infection (with a target of having at least 30% of the sample made up of patients with compensated cirrhosis) were randomly assigned in a 1:1 ratio to receive sofosbuvir-velpatasvir-voxilaprevir or matched placebo for 12 weeks; randomization was stratified according to cirrhosis status. The investigators, patients, and study personnel were unaware of the study-group assignments for patients with genotype 1 infection until after the post-treatment week 4 visit. All patients who were infected with HCV of other genotypes or an indeterminate genotype, regardless of whether they had cirrhosis, were enrolled in the sofosbuvir-velpatasvir-voxilaprevir group, since patients with non-genotype 1 infection comprise a comparatively small proportion of those who have virologic failure after treatment with a DAA-based regimen. Patients received either a fixed-dose combination tablet containing 400 mg of sofosbuvir, 100 mg of velpatasvir, and 100 mg of voxilaprevir or a matching placebo tablet, administered orally once daily for 12 weeks. Patients who were randomly assigned to the placebo group were eligible for 12 weeks of subsequent treatment with sofosbuvir-velpatasvir-voxilaprevir.
In POLARIS-4, patients were enrolled at 101 sites in the United States, Canada, New Zealand, Australia, France, Germany, and the United Kingdom from January through May 2016. In this open-label trial, patients with HCV genotype 1, 2, or 3 infection were assigned in a 1:1 ratio to receive either sofosbuvir-velpatasvir-voxilaprevir or sofosbuvir-velpatasvir once daily for 12 weeks; randomization was stratified according to HCV genotype and cirrhosis status. Patients who were infected with HCV of any other genotype were assigned to receive sofosbuvir-velpatasvir-voxilaprevir for 12 weeks.
Assessments
For both trials, screening assessments included measurement of the serum HCV RNA level, IL28B genotyping, and standard laboratory and clinical testing. HCV RNA levels were measured with the use of the COBAS AmpliPrep/COBAS TaqMan HCV Quantitative Test, version 2.0, with a lower limit of quantification of 15 IU per milliliter. IL28B genotype was determined by polymerase chain reaction amplification of the single-nucleotide polymorphism rs12979860, with the use of TaqMan MGB probes.
The Abbott RealTime HCV genotype II assay was used to determine HCV genotype at screening. HCV genotype and subtype were subsequently determined by analysis of NS3, NS5A, and NS5B sequences obtained by deep sequencing with the use of the Basic Local Alignment Search Tool (BLAST); these results were used in the analyses.
Deep sequencing of the NS3, NS5A, and NS5B coding regions was performed on samples obtained from all the patients at baseline and from those patients with virologic failure at the time of failure. Sequences that were obtained at the time of virologic failure were compared with sequences from baseline samples to detect resistance-associated substitutions that arose in association with treatment. We report resistance-associated substitutions that were present in more than 15% of sequence reads.
End Points
For both trials, the primary efficacy end point was a sustained virologic response, defined as a serum HCV RNA level lower than 15 IU per milliliter 12 weeks after the end of treatment in patients who were enrolled and received at least one dose of active treatment or placebo. Patients whose HCV RNA levels were not assessed at 12 weeks after treatment for any reason were classified as not having had a sustained virologic response, with the exception of those who had an HCV RNA level lower than 15 IU per milliliter both before and after post-treatment week 12; a sustained virologic response at post-treatment week 12 was imputed for these patients. The primary safety end point was the proportion of patients who stopped taking active treatment or placebo prematurely owing to adverse events.
The secondary efficacy end points were the percentage of patients with an HCV RNA level lower than 15 IU per milliliter at 4 and 24 weeks after the end of treatment, the percentage of patients with an HCV RNA level lower than 15 IU per milliliter during treatment, the change in HCV RNA level from baseline (day 1), and the proportion of patients with virologic failure (defined as a confirmed HCV RNA level of at least 15 IU per milliliter after two consecutive measurements showing HCV RNA levels lower than 15 IU per milliliter, or an increase in the HCV RNA level of more than 1 log10from the nadir during the treatment period). In addition, we report results for the secondary objective of an assessment of the emergence of viral resistance to the study drugs. Evaluation of other secondary objectives for POLARIS-1 — the characterization of the steady-state pharmacokinetics of sofosbuvir, velpatasvir, and voxilaprevir, and an evaluation of the efficacy and safety of deferred treatment with 12 weeks of sofosbuvir-velpatasvir-voxilaprevir in the placebo group — are beyond the scope of the current report.
Statistical Analysis
In both trials, the primary efficacy analysis was designed to test for the superiority of the rate of sustained virologic response achieved among patients receiving sofosbuvir-velpatasvir-voxilaprevir or sofosbuvir-velpatasvir over a performance goal of 85%, with the use of a two-sided exact one-sample binomial test at the 0.05 significance level for POLARIS-1 and at the 0.025 significance level for POLARIS-4, on the basis of the Bonferroni adjustment for two primary efficacy tests. The planned enrollment of 280 patients in the sofosbuvir-velpatasvir-voxilaprevir group in POLARIS-1 and of 205 patients in the sofosbuvir-velpatasvir-voxilaprevir group and 175 in the sofosbuvir-velpatasvir group in POLARIS-4 were calculated to provide more than 90% power to detect an advantage of 10 percentage points in the rate of sustained virologic response over the performance goal of 85%, which was based on the efficacy of the combination of sofosbuvir-velpatasvir-voxilaprevir in phase 2 trials. Confidence intervals for the efficacy rates according to subgroup were not adjusted for multiplicity. POLARIS-4 was not powered for a comparison between sofosbuvir-velpatasvir-voxilaprevir and sofosbuvir-velpatasvir, and no statistical comparisons between the groups were planned or performed. The results with regard to the secondary end points and objectives were summarized.
Results
Patients
Of the 520 patients who underwent screening for POLARIS-1, 416 were enrolled, and 415 began the active treatment or placebo (Table S1 and Fig. S1 in the Supplementary Appendix). In total, we enrolled 300 patients with HCV genotype 1 infection, 5 with genotype 2 infection, 78 with genotype 3 infection, 22 with genotype 4 infection, 1 with genotype 5 infection, 8 with genotype 6 infection, and 1 with infection with HCV of unknown genotype. Of the 300 patients with HCV genotype 1 infection, 150 were randomly assigned to the sofosbuvir-velpatasvir-voxilaprevir group, and 150 were assigned to the placebo group. The other 114 patients with non-genotype 1 HCV infection at screening were enrolled in the sofosbuvir-velpatasvir-voxilaprevir group. Of these patients, 1 with HCV genotype 4 infection never received treatment.
Of the 397 patients who underwent screening for POLARIS-4, 333 were enrolled and treated: 144 patients with HCV genotype 1 infection, 64 with genotype 2 infection, 106 with genotype 3 infection, and 19 with genotype 4 infection (Table S2 and Fig. S2 in the Supplementary Appendix). No patients with HCV genotype 5 or 6 infection were enrolled. In total, 163 patients were randomly assigned to receive sofosbuvir-velpatasvir-voxilaprevir (78 patients with genotype 1 infection, 31 with genotype 2 infection, and 54 with genotype 3 infection), and 151 were assigned to receive sofosbuvir-velpatasvir (66 with genotype 1 infection, 33 with genotype 2 infection, and 52 with genotype 3 infection). Per protocol, all 19 patients with HCV genotype 4 infection were enrolled in the sofosbuvir-velpatasvir-voxilaprevir group.
The demographic and baseline clinical characteristics of the patients in both trials are shown in Table 1 in each of the three active-treatment groups, the percentage of patients with compensated cirrhosis was 46%. In POLARIS-1, the most common NS5A inhibitors used in previous unsuccessful treatment were ledipasvir (55% of patients), daclatasvir (23%), and ombitasvir (13%). In POLARIS-4, 85% of patients had received sofosbuvir as a part of previous unsuccessful treatment.
Efficacy
POLARIS-1
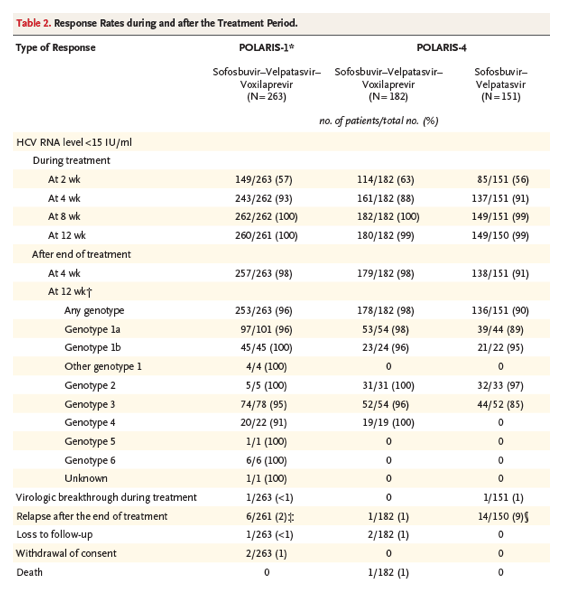
Among the patients who had previously been treated with a regimen containing an NS5A inhibitor, the overall rate of sustained virologic response in the sofosbuvir-velpatasvir-voxilaprevir group was 96% (95% confidence interval [CI], 93 to 98), which was significantly superior to the prespecified performance goal of 85% (P<0.001) (Figure 1 and Table 2). Of the 253 patients with a sustained virologic response at week 12 after treatment, 249 returned for the post-treatment week 24 visit. All 249 patients had a sustained virologic response at that time. None of the patients who received placebo had a sustained virologic response. The HCV RNA levels and changes from baseline at each visit through the end of treatment are provided in Table S3 in the Supplementary Appendix.
The rates of sustained virologic response were 96% (95% CI, 90 to 99) among patients with HCV genotype 1a infection, 100% (95% CI, 92 to 100) among those with genotype 1b infection, 100% (95% CI, 48 to 100) among those with genotype 2 infection, 95% (95% CI, 87 to 99) among those with genotype 3 infection, 91% (95% CI, 71 to 99) among those with genotype 4 infection, and 100% (95% CI, 54 to 100) among those with genotype 6 infection. The single patient with HCV genotype 5 infection had a sustained virologic response. Overall, the rate of sustained virologic response was 99% (95% CI, 95 to 100) among patients who did not have cirrhosis and 93% (95% CI, 87 to 97) among those who had cirrhosis. Of the 56 patients with genotype 3 infection and cirrhosis, 52 had a sustained virologic response (93%; 95% CI, 83 to 98).
Among the 263 patients who received sofosbuvir-velpatasvir-voxilaprevir, 10 did not have a sustained virologic response. Of these 10 patients, 7 had virologic failure: 1 (<1%) had virologic breakthrough during treatment, and 6 (2%) had virologic relapse after the end of treatment. The patient with virologic breakthrough had low plasma concentrations of GS-331007 (the chief sofosbuvir metabolite), velpatasvir, and voxilaprevir at weeks 8 and 12, which was suggestive of nonadherence. Two patients receiving sofosbuvir-velpatasvir-voxilaprevir withdrew consent, one after completing treatment and having a virologic response 4 weeks after the end of treatment and another after taking four doses of study drug (as described below). One other patient was lost to follow-up after week 8 of treatment. The characteristics of the 7 patients who had virologic failure are provided in Table S5 in the Supplementary Appendix.
POLARIS-4
Among the patients who had previously been treated with a regimen containing any DAA except an NS5A inhibitor, the overall rate of sustained virologic response was 98% (95% CI, 95 to 99) among those who received sofosbuvir-velpatasvir-voxilaprevir, which was significantly superior to the prespecified performance goal of 85% (P<0.001) (Table 2 and Figure 1). The rate of sustained virologic response among the patients who received sofosbuvir-velpatasvir was 90% (95% CI, 84 to 94), which was not significantly superior to the prespecified performance goal of 85% (P=0.09). The HCV RNA levels and changes from baseline at each study visit through the end of treatment are provided in Table S4 in the Supplementary Appendix.
Among patients without cirrhosis, the rate of sustained virologic response was 98% among those receiving sofosbuvir-velpatasvir-voxilaprevir and 94% among those receiving sofosbuvir-velpatasvir, as compared with 98% and 86%, respectively, among patients with cirrhosis. Table 2 shows rates of sustained virologic response according to HCV genotype. Of the 177 patients in the sofosbuvir-velpatasvir-voxilaprevir group and the 136 patients in the sofosbuvir-velpatasvir group who had a sustained virologic response at post-treatment week 12, a total of 173 and 133 patients, respectively, returned for the post-treatment week 24 visit, and all the patients had a sustained virologic response at that time. One patient in the sofosbuvir-velpatasvir-voxilaprevir group did not attend the post-treatment week 12 visit; however, this patient returned at post-treatment week 24 and had an HCV RNA level lower than 15 IU per milliliter. In accordance with the prespecified analysis plan, a sustained virologic response at post-treatment week 12 was imputed for this patient.
Among the 333 patients who were treated in POLARIS-4, 19 did not have a sustained virologic response — 4 patients (3%) in the sofosbuvir-velpatasvir-voxilaprevir group and 15 patients (10%) in the sofosbuvir-velpatasvir group. Of the 4 patients in the sofosbuvir-velpatasvir-voxilaprevir group who did not have a sustained virologic response, 1 (1%) had a virologic relapse by week 4 of follow-up, 1 died (see below), and 2 were lost to follow-up. Among the 15 patients in the sofosbuvir-velpatasvir group who did not have a sustained virologic response, 14 (9%) had a relapse after completing treatment and 1 (1%) had virologic breakthrough during treatment. Eight of the 14 patients who had a relapse had HCV genotype 3a infection, 5 had genotype 1a infection, and 1 had genotype 1b infection (Table S6 in the Supplementary Appendix).
Viral Resistance Testing
Among the 248 patients who received sofosbuvir-velpatasvir-voxilaprevir in POLARIS-1 and for whom viral sequence data were available, 205 (83%) had viral substitutions associated with resistance to NS3 inhibitors or NS5A inhibitors at baseline that were present in at least 15% of sequence reads. Of these patients, 97% (199 of 205) had a sustained virologic response, as compared with 98% of patients without resistance-associated substitutions at baseline (Table S7 in the Supplementary Appendix). Among the 6 patients who had a relapse, 1 patient with HCV genotype 4 infection had development of the NS5A Y93H resistance-associated substitution (Table S5 in the Supplementary Appendix).
In POLARIS-4, 49% of enrolled patients had baseline viral substitutions associated with resistance to NS3 inhibitors or NS5A inhibitors. The rates of sustained virologic response among patients for whom viral sequence data were available and who received sofosbuvir-velpatasvir-voxilaprevir for 12 weeks was 100% (83 of 83) among those with baseline resistance-associated substitutions and 99% (85 of 86) among those without baseline resistance-associated substitutions, as compared with 90% (63 of 70) and 89% (67 of 75), respectively, among those with and those without resistance-associated substitutions in the sofosbuvir-velpatasvir group (Table S8 in the Supplementary Appendix). The single patient in the sofosbuvir-velpatasvir-voxilaprevir group who had a relapse did not have any resistance-associated substitutions at either baseline or the time of relapse. Among the 14 patients in the sofosbuvir-velpatasvir group who had a relapse, 11 had resistance-associated substitutions, most of which were in the NS5A gene at amino acid position 93 (Table S6 in the Supplementary Appendix).
Safety
One patient who received sofosbuvir-velpatasvir-voxilaprevir in POLARIS-1 discontinued treatment prematurely because of an adverse event. This patient, a 59-year-old woman with a history of hypertension, started therapy with ramipril on study day 11 and discontinued sofosbuvir-velpatasvir-voxilaprevir treatment on day 12 because of angioedema. Three patients who received placebo in POLARIS-1 discontinued because of adverse events. In POLARIS-4, 1 patient, a 63-year-old woman in the sofosbuvir-velpatasvir group, discontinued treatment on study day 49 because of worsening of headache. None of the patients in POLARIS-4 who received sofosbuvir-velpatasvir-voxilaprevir discontinued treatment prematurely because of adverse events.
A total of seven serious adverse events occurred among 5 patients (2%) who received sofosbuvir-velpatasvir-voxilaprevir in POLARIS-1 (Table 3, and Table S9 in the Supplementary Appendix). In the placebo group, 7 patients had one serious adverse event each. In POLARIS-4, 4 patients (2%) receiving sofosbuvir-velpatasvir-voxilaprevir and 4 patients (3%) receiving sofosbuvir-velpatasvir had one serious adverse event each (Table S10 in the Supplementary Appendix). Across both trials, no single serious adverse event occurred in more than 1 patient. One patient, a 61-year-old man, died from an illicit drug overdose 2 days after completing the 12 weeks of treatment with sofosbuvir-velpatasvir-voxilaprevir.
In POLARIS-1, 78% of patients who received sofosbuvir-velpatasvir-voxilaprevir had adverse events, as compared with 70% of patients who received placebo (Table 3). The most common events in the sofosbuvir-velpatasvir-voxilaprevir group were headache (25% of patients), fatigue (21%), diarrhea (18%), and nausea (14%), and the most common events in the placebo group were fatigue (20%), headache (17%), diarrhea (12%), and dizziness (9%). In POLARIS-4, the incidence of adverse events was 77% among patients who received sofosbuvir-velpatasvir-voxilaprevir and 74% among those who received sofosbuvir-velpatasvir. The most common events among patients who received sofosbuvir-velpatasvir-voxilaprevir were headache (27%), fatigue (24%), and diarrhea (20%), and the most common events among those who received sofosbuvir-velpatasvir were headache (28%), fatigue (28%), and nausea (8%). The majority of events of diarrhea were mild in severity; the incidence of grade 2 diarrhea was low (1 to 3%). There were no events of grade 3 or 4 diarrhea. The grade 3 and 4 adverse events in POLARIS-1 and POLARIS-4 are presented in Tables S11 and S12 in the Supplementary Appendix.
The incidence of grade 3 and 4 laboratory abnormalities was 5% and 2%, respectively, among patients receiving sofosbuvir-velpatasvir-voxilaprevir in POLARIS-1, as compared with 12% and 2% among those receiving placebo (Table 3, and Table S13 in the Supplementary Appendix). In POLARIS-4, the incidence of grade 3 and 4 laboratory abnormalities was 5% and less than 1%, respectively, among patients receiving sofosbuvir-velpatasvir-voxilaprevir, as compared with 6% and 1% among patients receiving sofosbuvir-velpatasvir (Table 3, and Table S14 in the Supplementary Appendix). None of the grade 3 and 4 elevations in lipase and creatine kinase levels were accompanied by clinical evidence of pancreatitis or myopathy, respectively.
Discussion
In these international phase 3 trials, 12 weeks of treatment with sofosbuvir-velpatasvir-voxilaprevir resulted in high rates of sustained virologic response among patients with and patients without compensated cirrhosis who had HCV of any genotype and who had not had a sustained virologic response after previous treatment with regimens containing DAAs, including NS5A inhibitors. This population of patients has been underrepresented in clinical trials and has limited retreatment options. It should be noted that these trials involved only patients who had observed virologic failure after having completed previous treatment; it excluded those who did not have a sustained virologic response as a result of nonadherence or those who had discontinued previous treatment owing to adverse events.
Because of their potency, NS5A inhibitors have been a common component of DAA regimens. Substitutions in the viral genome that confer resistance to NS5A inhibitors, unlike those that confer resistance to NS3 protease inhibitors and NS5B polymerase inhibitors, appear to maintain the viability of the virus after unsuccessful treatment with an NS5A inhibitor-containing regimen and have been shown to have an effect on the rate of sustained virologic response in previous studies of DAA regimens.19 As expected, POLARIS-1 and POLARIS-4 enrolled a substantial number of patients who had resistance-associated viral substitutions at baseline, but the presence of such substitutions had no discernible effect on the rates of sustained virologic response with sofosbuvir-velpatasvir-voxilaprevir.
The incidence of adverse events among the patients who received sofosbuvir-velpatasvir-voxilaprevir was generally similar to the rates among patients who received placebo in POLARIS-1 and sofosbuvir-velpatasvir in POLARIS-4, except that more patients who received sofosbuvir-velpatasvir-voxilaprevir in POLARIS-1 had headache and more patients who received sofosbuvir-velpatasvir-voxilaprevir in both trials had mild-to-moderate nausea and diarrhea, which are known effects of some NS3-NS4A protease inhibitors.20 No patient interrupted treatment or discontinued treatment prematurely as a result of these events.
The generalizability of these results may be limited by the small numbers of patients in some subpopulations, including those with genotype 3 infection and cirrhosis and those infected with rarer genotypes, as well as by the fact that some patients did not receive previous treatment with commercially available regimens. Another limitation that affects the generalizability of these trials is the small number of retreated patients who had treatment failure with the more recently approved HCV regimens that include velpatasvir or elbasvir. The results also cannot be generalized to patients who were excluded from the trials, such as those coinfected with hepatitis B virus or human immunodeficiency virus and those with decompensated cirrhosis.
In conclusion, these results show that daily treatment with the single-tablet regimen of sofosbuvir-velpatasvir-voxilaprevir for 12 weeks is highly effective for patients infected with HCV of any genotype, with or without compensated cirrhosis, who did not have a sustained virologic response after treatment with DAA-based regimens, including NS5A inhibitors.
| |
| |
| |
|
|
|