| |
Doravirine versus ritonavir-boosted darunavir in antiretroviral-naive adults with HIV-1 (DRIVE-FORWARD): 48-week results of a randomised, double-blind, phase 3, non-inferiority trial
|
| |
| |
Download the PDF here
Download the PDF here
Lancet HIV March 25 2018 - Jean-Michel Molina, Kathleen Squires, Paul E Sax, Pedro Cahn, Johan Lombaard, Edwin DeJesus, Ming-Tain Lai, Xia Xu, Anthony Rodgers, Lisa Lupinacci, Sushma Kumar, Peter Sklar, Bach-Yen Nguyen, George J Hanna, Carey Hwang, for the DRIVE-FORWARD Study Group
CROI 2018: Similar Efficacy and Safety By Subgroup in DRIVE-AHEAD: DOR/3TC/TDF vs EFV/FTC/TDF - (03/06/18)
Summary
Background
Doravirine is a novel non-nucleoside reverse transcriptase inhibitor (NNRTI) with a pharmacokinetic profile supporting once-daily dosing, and potent in-vitro activity against the most common NNRTI-resistant HIV-1 variants. We compared doravirine with ritonavir-boosted darunavir, when both were given with two nucleoside reverse transcriptase inhibitors (NRTIs), in adults with previously untreated HIV-1 infection.
Methods
In this randomised, controlled, double-blind, multicentre, non-inferiority trial, adults with HIV-1 infection were screened and enrolled at 125 clinical centres in 15 countries. Eligible participants (aged ≥18 years) were naive to antiretroviral therapy with plasma HIV-1 RNA of at least 1000 copies per mL at screening. Participants who had previously been treated for a viral infection other than HIV-1, those taking immunosuppressive drugs, and individuals with active acute hepatitis were excluded. Participants were randomly assigned (1:1) via an interactive voice and web response system to receive oral doravirine 100 mg or darunavir 800 mg plus ritonavir 100 mg once daily, with two investigator-selected NRTIs (tenofovir and emtricitabine or abacavir and lamivudine) for up to 96 weeks. Randomisation was stratified by HIV-1 RNA measurements at screening (≤100 000 vs >100 000 copies per mL) and the NRTI pair. Study participants, funding institution staff, investigators, and study site personnel were masked to treatment group assignment. The primary efficacy endpoint was the proportion of participants achieving HIV-1 RNA of less than 50 copies per mL at week 48 defined by the US Food and Drug Administration snapshot algorithm, with non-inferiority established if the lower bound of the two-sided 95% CI for the treatment difference (doravirine minus darunavir) was greater than -10 percentage points. All participants who received at least one dose of study drug were included in the primary efficacy and safety analyses. This trial is active, but not recruiting, and is registered with ClinicalTrials.gov, number NCT02275780.
Findings
Between Dec 1, 2014, and Oct 20, 2015, 1027 participants were screened for eligibility, of whom 769 participants were randomly assigned to treatment (385 with doravirine and 384 with ritonavir-boosted darunavir). 56 participants discontinued treatment in the doravirine group compared with 71 in the darunavir group, mostly due to loss to follow-up. 383 participants who received doravirine and 383 who received darunavir were included in the primary efficacy analyses. At week 48, 321 (84%) participants in the doravirine group and 306 (80%) in the darunavir group achieved plasma HIV-1 RNA of less than 50 copies per mL (difference 3⋅9%, 95% CI -1⋅6 to 9⋅4), indicating non-inferiority of the doravirine regimen. The most common study drug-related adverse events were diarrhoea (21 [5%] of 383 participants in the doravirine group and 49 [13%] of 383 participants in the darunavir group), nausea (25 [7%] vs 29 [8%]), and headache (23 [6%] vs ten [3%]). 18 participants (six [2%] of 383 participants in the doravirine group vs 12 [3%] of 383 participants in the darunavir group) discontinued treatment due to adverse events, which were considered drug-related in four (1%) participants in the doravirine group and 8 (2%) participants in the darunavir group. Serious adverse events occurred in 19 (5%) of 383 participants in the doravirine group and 23 (6%) of 383 in the darunavir roup, and were considered study-drug related in one (<1%) participant of each group.
Interpretation
In treatment-naive adults with HIV-1 infection, doravirine combined with two NRTIs might offer a valuable treatment option for adults with previously untreated HIV-1 infection.
Funding
Merck & Co.
Introduction
Although non-nucleoside reverse transcriptase inhibitors (NNRTIs) are important components of combination antiretroviral therapy for previously untreated HIV-1 infection, all available drugs in this class have disadvantages. Efavirenz is the preferred third drug for use in combination with tenofovir and emtricitabine in WHO guidelines, but in other guidelines (US Department of Health and Human Services, European AIDS Clinical Society [EACS], and British HIV Association [BHIVA] guidelines) it is included only as an alternative regimen because of CNS intolerance1 and possible association with suicidality.2 Rash is another common side-effect of efavirenz,3 and lipid abnormalities seem to be more common with efavirenz than with other NNRTIs.4 Rilpivirine has low antiviral efficacy in patients with high viral load5, 6 and therefore is not recommended for individuals with baseline HIV-1 RNA of more than 100 000 copies per mL or CD4 counts of less than 200 cells per μL because of the increased risk of virological failure.7 Additionally, rilpivirine requires dosing with food and should not be given with proton-pump inhibitors, which results in substantial lowering of rilpivirine plasma concentrations.7 Nevirapine is associated with serious dermatological and hepatic toxicity and should not be prescribed in men with CD4 counts of more than 400 cells per μL or in women with counts greater than 250 cells per μL.8 Etravirine is not approved for first-line treatment and requires twice-daily dosing.9 New NNRTI drugs without these limitations are needed.
Discussion
In this randomised, double-blind, multicentre, phase 3, non-inferiority trial doravirine was compared with darunavir, both in combination with two NRTIs, for the treatment of antiretroviral-naive adults with HIV-1. At week 48, the efficacy of doravirine was non-inferior to that of darunavir, with 84% of participants in the doravirine group and 80% of participants in the darunavir group achieving plasma HIV-1 RNA counts of less than 50 copies per mL. The efficacy of doravirine was similar to that of ritonavir-boosted darunavir in participants with baseline HIV-1 RNA of more than 100 000 copies per mL or with baseline CD4 counts of less than 200 cells per μL. The change in CD4 cell counts from baseline to week 48 was also similar in the two treatment groups.
The proportion of participants with HIV-1 RNA of less than 50 copies per mL in the doravirine group (84%) was similar to that observed for the NNRTIs rilpivirine and efavirenz in ECHO (83% and 83%)5 and THRIVE (86% and 82%).27 However, the proportion of participants with a virological response in the darunavir group (80%) was lower than reported in ARTEMIS (84%)28 and FLAMINGO (83%).29 This result might be associated with the proportion of participants who discontinued treatment with ritonavir-boosted darunavir for reasons other than poor efficacy (57 [15%] of 383 participants), which decreases the observed response rate found with the FDA snapshot algorithm.
Another possible reason for lower efficacy is the definition of PDVF used in this trial: specifically, discontinuation from the trial was required for participants with confirmed plasma HIV-1 RNA of more than 50 copies per mL after suppression to less than 50 copies per mL at any time during the trial. Other clinical trials have allowed participants to remain on study treatment despite meeting PDVF criteria or have used a higher, clinically relevant threshold for PDVF (ie, 200 copies per mL).29, 30 Most participants who met PDVF criteria in our study had HIV-1 RNA of less than 200 copies per mL and most participants with virological rebound had HIV-1 RNA of less than 100 copies per mL. If these participants had been allowed to continue in the trial, viral loads might have been suppressed again to less than 50 copies per mL in some participants. Notably, efficacy results for the darunavir group determined with the observed failure approach (86%) were more similar to results of other trials28, 29 of darunavir in treatment-naive participants with HIV-1 infection.
The efficacy observed across subgroups indicates that doravirine has similar efficacy to an approved preferred protease inhibitor that is part of the current recommended standard of care in EACS and BHIVA treatment guidelines. Although lower efficacy was observed in participants with high baseline HIV-1 RNA measurements or low pretreatment CD4 cell count in both treatment groups, these findings are consistent with previous reports31, 32 for other antiretroviral drugs, and were less pronounced for the doravirine group than for the darunavir group. In both treatment groups, efficacy was similar regardless of the NRTI component, which was tenofovir and emtricitabine in 668 (87%) of 769 participants.
The proportion of participants with PDVF was low: 5% in the doravirine group and 6% in the darunavir group. Among individuals with PDVF who had successful genotype and phenotype testing, no primary genotypic resistance mutations or phenotypic resistance to any study drug was identified in either treatment group. Of 127 participants who discontinued early, one individual in the doravirine group developed both genotypic and phenotypic resistance to doravirine and emtricitabine. Thus, the overall proportion of participants who developed resistance to doravirine was 0⋅3% (one of 383), which is lower than reported for other NNRTIs in recent trials (eg, 1⋅0-2⋅3% for efavirenz after 48 weeks).33, 34 However, additional data are needed to confirm these findings because resistance was assessed in only a few participants with low viral loads. Additionally, cross-resistance to other NNRTIs requires investigation.
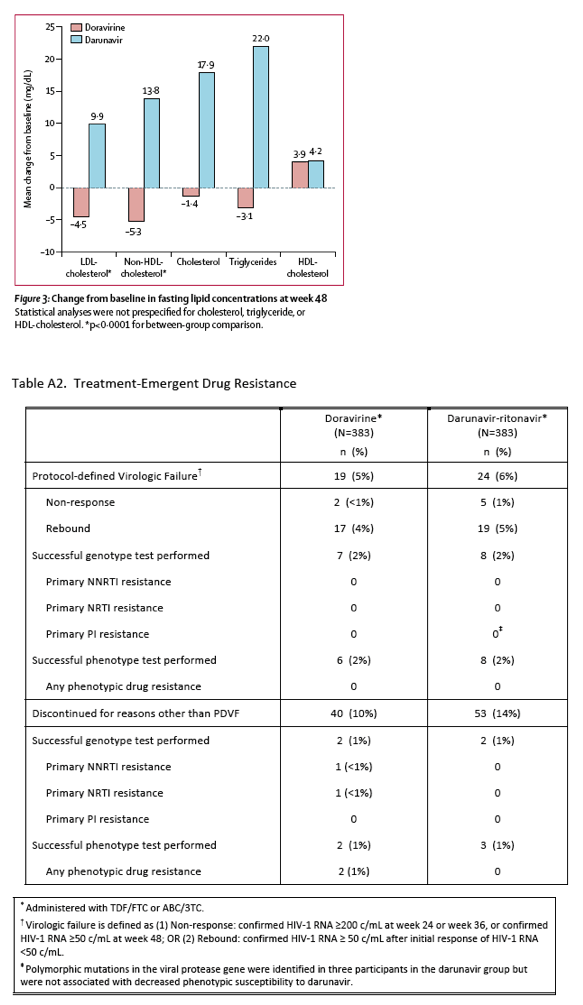
Doravirine was generally well tolerated, with few discontinuations because of adverse events (2%) and only one drug-related serious adverse event (1%). We found no clinically relevant differences in incidence of specific adverse events between treatment groups, with the exception of a higher incidence of diarrhoea in the darunavir group (22% vs 14%), which is consistent with the known safety profile of ritonavir-boosted darunavir. The effect of doravirine on fasting lipid concentrations was superior to that of ritonavir-boosted darunavir, as shown by significant between-treatment differences for the mean change from baseline in LDL-cholesterol and non-HDL-cholesterol concentrations.
The incidences of rash and neuropsychiatric adverse events in the doravirine group were similar to those in the darunavir group, and most of these events were of mild intensity. Only two participants discontinued treatment with doravirine because of rash, and no participants in the trial discontinued due to a neuropsychiatric adverse event. Compared with other NNRTIs, the incidences of rash and neuropsychiatric events in this study were similar to those observed with rilpivirine and lower than those observed with efavirenz in the ECHO and THRIVE studies.5, 27 In a direct comparison with efavirenz, the proportion of participants with neuropsychiatric events was significantly lower in the doravirine group at week 8 (22% vs 44%)22 and week 24 (27% vs 46%).23 Preliminary results from the ongoing DRIVE-AHEAD35 study show a superior neuropsychiatric profile for the fixed combination of doravirine, lamivudine, and tenofovir disoproxil fumarate compared with efavirenz, emtricitabine, and tenofovir.
The ritonavir-boosted darunavir regimen was chosen as the comparator in this study because it is among the recommended first-line drugs in multiple HIV treatment guidelines. Because darunavir and ritonavir are not coformulated, study participants were required to take four pills daily to conceal treatment assignments. This led to challenges for recruitment and retention and might have contributed to the higher rate of loss to follow-up and participant withdrawal than in other trials. Although the resistance profile of doravirine seems encouraging since only one participant developed genotypic and phenotypic resistance to doravirine, more clinical data for doravirine are needed. Ongoing and future studies will provide further insight into the resistance profile of doravirine. Another limitation of this study is the low number of women (121 [16%]) and participants aged older than 65 years (1%) enrolled in the trial.
In summary, we found that the antiretroviral efficacy of doravirine was non-inferior to that of ritonavir-boosted darunavir, as assessed by the proportion of participants achieving HIV-1 RNA less than 50 copies per mL at week 48. The antiviral response rates were similar in both treatment groups regardless of baseline factors, such as HIV-1 RNA of more than 100 000 copies per mL and CD4 counts of less than 200 cells per μL. Resistance to doravirine developed in one participant, who was discontinued from the trial by the investigator because of non-compliance. Doravirine was generally well tolerated during 48 weeks of treatment and had a favourable safety and lipid profile compared with darunavir and ritonavir. For people with HIV, doravirine presents another treatment option, with broad efficacy that is similar to two well established antiretroviral drugs (darunavir and efavirenz), a unique resistance profile, excellent tolerability, a superior neuropsychiatric profile compared with efavirenz, and a superior lipid profile compared with ritonavir-boosted darunavir.
Research in context
Evidence before this study
We searched PubMed from inception to March 31, 2017, for clinical trials that mentioned doravirine or MK-1439 and found two previous clinical trials in adults with HIV-1. In a short-term monotherapy study in treatment-naive men with HIV-1 infection, doravirine 25 mg or 200 mg once daily for 7 days had robust antiviral activity, without evidence of viral resistance, and was generally well tolerated. In a phase 2, dose-ranging study in treatment-naive adults, doravirine 100 mg once daily administered with tenofovir disoproxil fumarate and emtricitabine was efficacious and well tolerated, with significantly fewer neuropsychiatric adverse events than efavirenz. To date, no studies have compared doravirine with a protease inhibitor for the treatment of HIV-1 infection.
Added value of this study
To the best of our knowledge, this is the first randomised, controlled, phase 3 trial of the safety and efficacy of doravirine 100 mg for HIV-1 infection. We found that in treatment-naive adults with HIV-1 infection, the antiretroviral efficacy of doravirine was non-inferior to that of ritonavir-boosted darunavir when given with two nucleoside reverse transcriptase inhibitors (NRTIs), as assessed by the proportion of participants who had plasma HIV-1 RNA of less than 50 copies per mL at week 48. Antiretroviral responses were similar in the doravirine and ritonavir-boosted darunavir groups regardless of baseline factors (eg, HIV-1 RNA >100 000 copies per mL and CD4 counts of <200 cells per μL). One participant developed resistance to doravirine and was discontinued by the investigator for non-compliance. Doravirine was generally well tolerated up to 48 weeks of treatment.
Implications of all the available evidence
The safety and efficacy profiles of doravirine observed in this study support and supplement the findings of previous studies and suggest that doravirine 100 mg once daily, given in combination with two NRTIs, might offer a valuable treatment option for adults with previously untreated HIV-1 infection.
Doravirine is a novel NNRTI with potent antiviral activity against wild-type HIV-1 (half maximal inhibitory concentration [IC50] 12 nM in the presence of 100% normal human serum) and variants with the most frequently transmitted NNRTI-resistance mutations (ie, Lys103Asn, Tyr181Cys, and Gly190Ala).10, 11 The in-vitro resistance profile of doravirine is distinct from other NNRTIs,12 and mutant viruses selected by efavirenz or rilpivirine, including those with reverse transcriptase Glu138Lys or Lys101Glu mutations, remain susceptible to doravirine.10, 12 Doravirine is a substrate for cytochrome P450 3A-mediated metabolism but is not thought to have drug interactions via major drug-metabolising enzymes or transporters.13 No clinically meaningful interactions were observed in healthy volunteers when doravirine was given with tenofovir disoproxil fumarate, atorvastatin, oral contraceptives, or pantoprazole.14, 15, 16, 17 In other phase 1 studies,18, 19, 20 the bioavailability of doravirine was not affected by food intake, age, sex, or moderate hepatic impairment.
In a short-term monotherapy study,21 doravirine 25 mg or 200 mg once daily for 7 days had antiviral activity and was generally well tolerated in treatment-naive men with HIV. In a phase 2 study22, 23 of treatment-naive adults, doravirine 100 mg once daily given with tenofovir disoproxil fumarate and emtricitabine was efficacious and well tolerated, with significantly fewer neuropsychiatric adverse events than efavirenz. On the basis of these promising results, we did a randomised, controlled, double-blind, phase 3, non-inferiority trial comparing doravirine with ritonavir-boosted darunavir, both given with two nucleoside reverse transcriptase inhibitors (NRTIs), in adults with previously untreated HIV-1 infection.
Results
Between Dec 1, 2014, and Oct 20, 2015, 1027 individuals were screened for participation in this study, and 769 were randomly assigned to treatment: 385 to the doravirine group and 384 to the darunavir group (figure 1). Of the 252 participants who did not meet eligibility criteria, 124 (49%) were excluded because they had one or more mutations associated with decreased susceptibility to any study drug, and 48 (19%) were excluded because they had screening plasma HIV-1 RNA of less than 1000 copies per mL or treatment for HIV-1 infection was not recommended on the basis of physician assessment. Of the 769 participants who were randomly assigned, 383 participants in the doravirine group and 383 participants in the darunavir received at least one dose of study drug and were included in the week 48 analyses. The last follow-up visit included in this publication was on Sept 29, 2016. Overall, 127 (17%) of 766 participants had discontinued study treatment by week 48 (56 [15%] of 383 participants in the doravirine group; 71 [19%] of 383 participants in the darunavir group). In both groups, the most common reason for early discontinuation was loss to follow-up (17 [4%] of 383 participants in the doravirine group; 19 [5%] of 383 participants in the darunavir group).
The median age of the treated population was 33 years (IQR 27-42) and 760 (99%) participants were aged younger than 65 years. The treated population included 645 (84%) men and 121 (16%) women, of whom 560 (73%) were white, 73 (10%) had previously been diagnosed with AIDS (as reported by the investigator), and 538 (70%) had subtype B HIV-1 infection (table 1). At baseline, 22% of the doravirine group and 19% of the ritonavir-boosted darunavir group had plasma HIV-1 RNA of more than 100 000 copies per mL, with 17 (4%) and 12 (3%), respectively, exceeding 500 000 copies per mL.
At week 48, 321 (84%) of 383 participants in the doravirine group and 306 (80%) of 383 participants in the darunavir group had plasma HIV-1 RNA of less than 50 copies per mL (difference 3⋅9%, 95% CI -1⋅6 to 9⋅4; table 2, figure 2), showing non-inferiority of doravirine to darunavir. Similar results were obtained in the per-protocol analysis (appendix p 4). The full characterisation of virological outcomes at week 48 defined by the FDA snapshot algorithm was similar between the treatment groups (table 2). The proportion of participants with HIV-1 RNA of less than 50 copies per mL at each timepoint was similar between the treatment groups, with both groups reaching a plateau at week 24 (figure 2). Among the participants with HIV-1 RNA of 100 000 copies per mL or higher at baseline, 64 (81%) of 79 participants in the doravirine group and 55 (76%) of 72 participants in the darunavir group had plasma HIV-1 RNA of less than 50 copies per mL at week 48 (difference 3⋅0%, 95% CI -11⋅2 to 17⋅1; appendix p 5). In the small subgroup of participants with HIV-1 RNA of more than 500 000 copies per mL at baseline, 14 (82%) of 17 particpants in the doravrine group and six (50%) of 12 participants in the darunavir group had plasma HIV-1 RNA of less than 50 copies per mL at week 48 (difference 30⋅9%, 95% CI -4⋅1 to 65⋅9). Among participants with low CD4 count (<200 cells per μL) at baseline, 34 (83%) of 41 in the doravirine group and 44 (72%) of 61 in the darunavir group had HIV-1 RNA of less than 50 copies per mL at week 48 (difference 9⋅4% [95% CI -7⋅4 to 26⋅2]).
Results for the secondary virological endpoints were consistent with those for the primary endpoint (appendix p 4). Using the observed failure approach, at week 48, 321 (88%) of 364 participants in the doravirine group and 306 (86%) of 355 participants in the darunavir group achieved HIV-1 RNA of less than 50 copies per mL. At week 48, the mean change from baseline in CD4 cell counts was 193 per μL (95% CI 172 to 214) in the doravirine group and 186 per μL (168 to 204) in the darunavir group (mean difference 7⋅1 per μL, 95% CI -20⋅8 to 35⋅0).
19 (5%) of 383 participants in the doravirine group and 24 (6%) of 383 participants in the darunavir group had PDVF at week 48, which was because of virological rebound after an initial response in most cases: 17 (89%) of 19 participants in the doravirine group and 19 (79%) of 24 participants in the darunavir group. At the virological failure confirmation visit, 12 (63%) of 19 participants in the doravirine group and 14 (58%) of 24 participants in the darunavir group had plasma HIV-1 RNA of less than 200 copies per mL. Among the participants with virological rebound, nine (53%) of 17 participants in the doravirine group and ten (53%) of 19 participants in the darunavir and ritonavir group had HIV-1 RNA of less than 100 copies per mL.
Resistance testing was done in 15 of 43 participants with PDVF at week 48 (seven participants in the doravirine group; eight in the darunavir group; appendix p 7). Of the remaining 28 participants with PDVF, 24 were not tested because they had HIV-1 RNA of less than 400 copies per mL (11 participants in the doravirine group; 13 in the darunavir group), two participants in the darunavir group had samples collected after the data cutoff, and two did not have samples collected because of site error (one participant in both groups). In the doravirine group, no genotypic mutations associated with resistance to doravirine were identified and no phenotypic resistance to doravirine was observed. In the darunavir group, polymorphic mutations in the viral protease gene were identified in three participants (with no decrease in phenotypic susceptibility to darunavir; appendix p 6). No primary genotypic resistance mutations or phenotypic resistance to emtricitabine, tenofovir, abacavir, or lamivudine were detected in any participant of either treatment group.
93 participants discontinued study treatment early for reasons other than PDVF, 40 (10%) of 383 participants in the doravirine group and 53 (14%) of 383 participants in the darunavir group; two (1%) participants in the doravirine group and three (1%) in the darunavir group had samples with sufficient HIV-1 RNA for resistance testing (>400 copies per mL) at the time of discontinuation (appendix p 6). One participant in the doravirine group, who discontinued treatment because of non-compliance at week 24, developed resistance to doravirine (reverse transcriptase Val106Ile, His221Tyr, and Phe227Cys mutations and IC50 97 times higher than wild-type virus IC50) and emtricitabine (reverse transcriptase Met184Val mutation). This participant did not meet the criteria for PDVF because no confirmation sample was collected. Another participant in the doravirine group, who discontinued treatment due to rash at week 2, was identified as phenotypically resistant (IC50 2⋅8 times higher than wild-type virus). However, no resistance mutations to doravirine or other NNRTIs were identified, and the IC50 increase was minimal when compared with the 2⋅2 times increase in IC50 identified in this participant at baseline.
Clinical adverse events were reported by 307 (80%) of 383 participants in the doravirine group and 300 (78%) of 383 participants in the darunavir group (table 3) and were considered related to study therapy in 117 (31%) participants and 123 (32%) participants, respectively. The most common drug-related adverse events were diarrhoea (21 [5%] of 383 participants in the doravirine group and 49 [13%] of 383 participants in the darunavir group), nausea (25 [7%] vs 29 [8%]), and headache (23 [6%] vs ten [3%]). These events were also the most commonly reported adverse events overall, regardless of association with study medication. With the exception of the higher incidence of diarrhoea in the darunavir group than the doravirine group, no clinically relevant differences were identified between treatment groups in the incidence of specific drug-related adverse events. The incidence of skin rash and neuropsychiatric events was similar between the treatment groups (table 3). Most cases of rash were mild (21 participants in the doravirine group; 27 participants in the darunavir group). Three participants discontinued treatment because of rash: two in the doravirine group (one moderate and one severe) and one (moderate) in the darunavir group. None of the neuropsychiatric events resulted in treatment discontinuation, and suicidal behaviour was not reported in either group.
19 (5%) of 383 participants in the doravirine group and 23 (6%) of 383 participants in the darunavir group had serious adverse events, but these were considered drug-related in only one participant (<1%) in each group: one participant in the doravirine group had nausea and vomiting that resolved after 4 days without dose interruption or modification, and one participant in the darunavir group had peripheral oedema and was withdrawn from the study, but recovered after 1⋅2 months. One participant (aged 41 years) in the doravirine group with history of tuberculosis, seizures, hypersensitivity, and hypertension, died after about 7 months in the study of unknown causes, which were not thought to be related to study therapy. On day 222 (the last reported dose), the patient reported dizziness and subsequently collapsed and died in his home. At the time of death, mild muscle spasms, mild cough, and mild diarrhoea were ongoing, but no other deterioration of health was reported before death.
Six (2%) of 383 participants in the doravirine group and 12 (3%) of 383 participants in the darunavir group discontinued the study due to adverse events, which were considered drug-related in four (1%) participants in the doravirine group (nausea [n=1], abdominal pain and nausea [n=1], and rash [n=2]) and eight (2%) participants in the darunavir and ritonavir group (abdominal pain and hiatus hernia [n=1]; abdominal pain, flatulence, and nausea [n=1]; diarrhoea [n=1]; increased alanine transaminase and aspartate transaminase [n=1]; increased alanine transaminase, increased aspartate aminotransferase, and creatine phosphokinase [n=1]; peripheral oedema [n=1]; pyrexia [n=1]; and rash [n=1]). The Kaplan-Meier product-limit estimates revealed a smaller risk of discontinuation as a result of adverse events in the doravirine group than the darunavir group (appendix p 7).
The incidence of grade 3 or 4 laboratory abnormalities were similar between the regimens (appendix p 8), with the exception of increases in LDL-cholesterol concentration (grade 3), which occurred in one (<1%) participant in the doravirine group compared with nine (3%) participants in the darunavir group (difference -2⋅5%, 95% CI -5⋅0 to -0⋅8). The mean change in LDL-cholesterol from baseline to week 48 was -4⋅5 mg/dL (SD 20⋅6) in the doravirine group versus 9⋅9 mg/dL (27⋅3) in the darunavir group (mean difference -14⋅6 mg/dL, 95% CI -18⋅2 to -11⋅1; p<0⋅0001, figure 3). The mean change in non-HDL-cholesterol was similar (mean difference -19⋅3 mg/dL, -23⋅3 to -15⋅4; p<0⋅0001, figure 3). Total cholesterol and triglyceride concentrations decreased slightly in the doravirine group and increased in the darunavir group, whereas HDL-cholesterol increased slightly in both groups (figure 3). Six (2%) of 383 participants in the doravirine group and four (1%) of 383 in the darunavir and group started lipid-lowering therapy during the first 48 weeks of the study.
A grade 3 increase in serum creatinine occurred in five (1%) of 383 participants in the doravirine group and ten (3%) of 383 in the darunavir group (appendix p 9). The mean change from baseline in serum creatinine over time ranged from 0⋅04 mg/dL (SD 0⋅07) to 0⋅07 mg/dL (0⋅09) in the doravirine group and from 0⋅05 mg/dL (0⋅09) to 0⋅06 mg/dL (0⋅10) in the darunavir group (appendix p 9). None of the study participants discontinued therapy due to suspected tenofovir-related renal disease.
|
|
| |
| |
|
|
|