| |
Nonalcoholic Steatohepatitis - Opportunities and Challenges-
A Randomized, Controlled Trial of the Pan-PPAR Agonist Lanifibranor in NASH
|
| |
| |
Download the PDF here
Download the PDF here
NEJM Oct 21 2021- Guadalupe Garcia-Tsao, M.D. From the Digestive Diseases Section, Yale University School of Medicine, New Haven, and the Digestive Diseases Section, Veterans Affairs Connecticut Healthcare System, West Haven - both in Connecticut.
Nonalcoholic steatohepatitis (NASH), the progressive form of nonalcoholic fatty liver disease, has superseded hepatitis C as the main cause of cirrhosis and the main reason for liver transplantation.1Contrary to hepatitis C, a liver-specific disorder caused by a single etiologic agent, NASH is a multifactorial, complex metabolic disorder that forms part of a systemic disease. Therefore, finding therapies (other than weight loss), identifying the target population, and defining response to therapy represent important challenges in NASH.
The diagnosis of NASH is established through a liver-biopsy sample that shows steatosis, lobular inflammation, and hepatocellular ballooning. NASH is classified into 5 stages on the basis of the extent of fibrosis: F0 indicates no fibrosis, F1 portal fibrosis, F2 periportal fibrosis, F3 bridging fibrosis, and F4 cirrhosis. In patients with noncirrhotic NASH, regulatory agencies consider the end points of resolution of NASH without worsening fibrosis and regression of fibrosis without worsening of NASH as surrogates of response.2 However, the relationship between histologic improvement and clinical outcomes has not been validated. Furthermore, biopsy sampling error3 and observer variability4 are substantial and may be partly responsible for an observed large placebo effect.
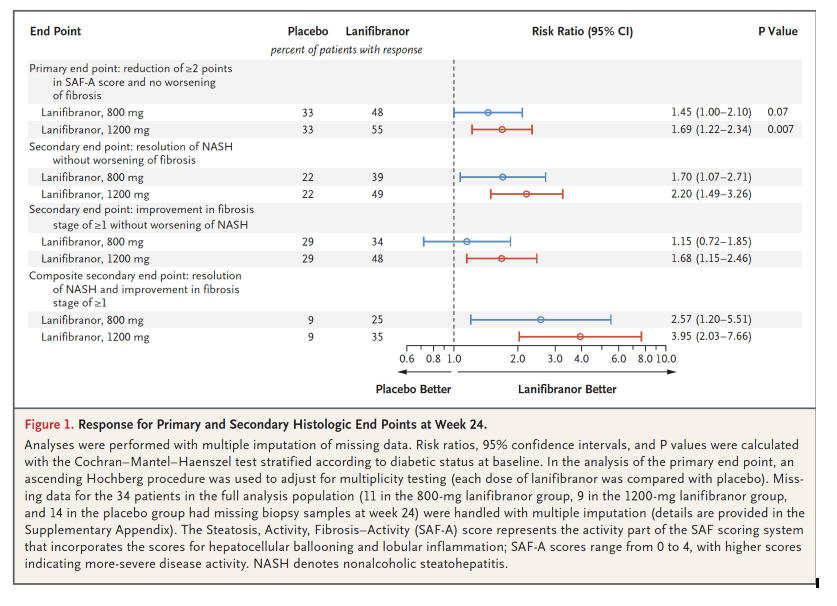
In this issue of the Journal, Francque et al.5 report the results of a phase 2b, multicenter, double-blind, randomized, placebo-controlled trial of lanifibranor, a pan-PPAR (peroxisome proliferator–activated receptor) agonist, in 275 patients with noncirrhotic NASH (76% of the patients had stage F2 or F3 fibrosis). The patients were randomly assigned to receive 1200 mg or 800 mg of lanifibranor or placebo daily for 24 weeks. The percentage of patients who met the primary end point - a decrease of at least 2 points in the Steatosis, Activity, Fibrosis (SAF)–Activity score, which ranges from 0 to 4 and focuses on ballooning and inflammation, without worsening of fibrosis - was significantly higher among those who received the 1200-mg dose of lanifibranor (55%), but not the 800-mg dose (48%), than among those who received placebo (33%). Resolution of NASH and improvement in fibrosis stage of at least 1, a composite end point that is unique to this trial, occurred in 35% of patients in the 1200-mg lanifibranor group, 25% of those in the 800-mg lanifibranor group, and 9% of those in the placebo group.
The next step in the regulatory pathway for lanifibranor is a phase 3, double-blind, randomized, placebo-controlled trial with clinical benefit as an end point. According to the Food and Drug Administration, the study population would comprise patients with NASH with stage F2 or F3 fibrosis, and clinical benefit would consist of “delaying progression to a composite endpoint including histological progression to F4, hepatic decompensation events (ascites, variceal hemorrhage, encephalopathy), change in MELD score from ≤12 to >15, liver transplant and all-cause mortality.”2 (The Model for End-Stage Liver Disease [MELD] score ranges from 6 to 40, with higher scores indicating a higher risk of death at 3 months.) Except for the end point regarding progression to cirrhosis, these end points are not different from those proposed for drugs that are being evaluated for compensated cirrhotic NASH.
The study by Sanyal et al.,6 also published in this issue of the Journal, describes the incidence and nature of clinical outcomes in one of the largest observational prospective studies of histologically characterized nonalcoholic fatty liver disease (75% of the patients had NASH). The study cohort, selected from the NASH Clinical Research Network, funded by the National Institute of Diabetes and Digestive and Kidney Diseases, included 1773 adults, of whom 1509 (85%) were White, 1237 (70%) had stage F0 to F2 fibrosis, 369 (21%) had stage F3 fibrosis, and 167 (9%) had stage F4 fibrosis. The patients were followed for a median of 4 years.
During the study period, there were 47 deaths from any cause (29 were among the patients with stage F3 or F4 fibrosis), 37 decompensation events (34 were among the patients with stage F3 or F4 fibrosis), and 9 cases of hepatocellular carcinoma (7 were among those with stage F3 or F4 fibrosis). Mortality was highest among the patients with stage F4 fibrosis (1.76 deaths per 100 person-years) and was lower among those with stage F3 fibrosis (0.89 deaths per 100 person-years) and even lower among those with stage F0 to F2 fibrosis (0.32 deaths per 100 person-years). Unlike retrospective cohort studies,7,8 this study did not confirm higher mortality among the patients with stage F2 fibrosis; 89% of the deaths that occurred among these patients were unrelated to liver disease. In the whole cohort, the occurrence of any new hepatic decompensation event was the only factor significantly associated with death, while nonhepatic new events (cardiac, declining kidney function, or extrahepatic cancer) were not significantly associated with death. Incident decompensation occurred predominantly among those with stage F4 fibrosis.
It follows that confirmatory clinical trials in NASH should focus on cirrhotic NASH (stage F4 fibrosis) by including patients with stage F3 or F4 fibrosis. End points in those with stage F3 fibrosis would be related to the prevention of progression to stage F4. End points in those with stage F4 fibrosis would be related to the prevention of decompensation or even to regression to stage F3. However, the need for liver biopsy in the selection of candidates and the assessment of outcomes is challenging.
A more feasible strategy - and one that would facilitate patient recruitment - would be to forgo liver biopsy and include only patients at high risk for decompensation. A combination of noninvasive markers that are currently used in clinical practice, including liver stiffness and routine laboratory tests (e.g., platelet count and albumin level), can identify patients who have clinically significant portal hypertension and thereby a higher likelihood of decompensation.9-11
In most cohort studies of cirrhosis,12,13 ascites and variceal hemorrhage are the most common decompensation events. Oddly, encephalopathy was the most common event in the cohort studied by Sanyal et al., a finding that raises the possibility of overdiagnosis of this clinically defined event and calls for adjudication of events. The use of the MELD score as an outcome in patients with NASH may not be as straightforward as it is for cirrhosis from other causes because of concurrent non–liver-related events, as shown in the study by Sanyal et al., in which nine patients with stage F0 to F2 fibrosis had a MELD score of 15 or higher (the cutoff used to list a patient for liver transplantation) at study entry but were receiving anticoagulants or had chronic kidney disease.
The study by Sanyal et al. had a modest number of outcomes, and this would translate into the need for very large sample sizes or a very long duration of follow-up, which are additional challenges in trials involving patients with NASH. The addition of other outcomes related to portal hypertension, such as the development or progression of gastroesophageal varices, and a study design that uses ordinal outcomes, in which clinical events are ordered by severity, would considerably reduce the sample size.14,15
The availability of new therapies that are effective in ameliorating the histologic features in NASH, as shown by Francque et al.,5 represents an invaluable opportunity. The challenge, as gleaned by Sanyal et al.,6 is in improving definitions of the patient population, outcomes, and clinical trial design that would prove that these therapies improve clinical outcomes.
-------------
Nonalcoholic Steatohepatitis - Opportunities and Challenges- A Randomized, Controlled Trial of the Pan-PPAR Agonist Lanifibranor in NASH - (10/25/21)
A Randomized, Controlled Trial of the Pan-PPAR Agonist Lanifibranor in NASH
S.M. Francque, P. Bedossa, V. Ratziu, Q.M. Anstee, E. Bugianesi, A.J. Sanyal, R. Loomba, S.A. Harrison, R. Balabanska, L. Mateva, N. Lanthier, N. Alkhouri, C. Moreno, J.M. Schattenberg, D. Stefanova Petrova, L. Vonghia, R. Rouzier, M. Guillaume, A. Hodge, M. Romero Gómez, P. Huot Marchand, M. Baudin, M. P. Richard, J. L. Abitbol, P. Broqua, J. L. Junien, and M.F. Abdelmalek, for the NATIVE Study Group*
Nonalcoholic steatohepatitis (NASH), a condition that results from a combination of adipose-tissue insulin resistance, adipocytokine imbalance, and systemic inflammation, is currently a major worldwide cause of chronic liver disease, contributing to cirrhotic morbidity, hepatocellular carcinoma and liver transplantation, worsening cardiovascular disease, and metabolic dysfunction.1-3Peroxisome proliferator–activated receptors (PPARs) are nuclear receptors with key regulatory functions in metabolism, inflammation, and fibrogenesis.4,5 In preclinical models, the indole sulfonamide lanifibranor (IVA337), a pan-PPAR agonist, improved insulin sensitivity and macrophage activation and reduced liver fibrosis and inflammatory gene expression with higher efficacy than single or dual PPAR agonists.6,7 Resolution of NASH and regression of fibrosis are currently considered to be likely surrogate outcomes for the prevention of progression to cirrhosis and associated complications.8 Here, we report the results of the NASH Trial to Validate IVA337 Efficacy (NATIVE), a phase 2b, double-blind, randomized, placebo-controlled trial evaluating the efficacy and safety of lanifibranor in patients with biopsy-proven, noncirrhotic NASH with severe disease activity.
Abstract
Background
Management of nonalcoholic steatohepatitis (NASH) is an unmet clinical need. Lanifibranor is a pan-PPAR (peroxisome proliferator–activated receptor) agonist that modulates key metabolic, inflammatory, and fibrogenic pathways in the pathogenesis of NASH.
Methods
In this phase 2b, double-blind, randomized, placebo-controlled trial, patients with noncirrhotic, highly active NASH were randomly assigned in a 1:1:1 ratio to receive 1200 mg or 800 mg of lanifibranor or placebo once daily for 24 weeks. The primary end point was a decrease of at least 2 points in the SAF-A score (the activity part of the Steatosis, Activity, Fibrosis [SAF] scoring system that incorporates scores for ballooning and inflammation) without worsening of fibrosis; SAF-A scores range from 0 to 4, with higher scores indicating more-severe disease activity. Secondary end points included resolution of NASH and regression of fibrosis.
Results
A total of 247 patients underwent randomization, of whom 103 (42%) had type 2 diabetes mellitus and 188 (76%) had significant (moderate) or advanced fibrosis. The percentage of patients who had a decrease of at least 2 points in the SAF-A score without worsening of fibrosis was significantly higher among those who received the 1200-mg dose, but not among those who received the 800-mg dose, of lanifibranor than among those who received placebo (1200-mg dose vs. placebo, 55% vs. 33%, P=0.007; 800-mg dose vs. placebo, 48% vs. 33%, P=0.07). The results favored both the 1200-mg and 800-mg doses of lanifibranor over placebo for resolution of NASH without worsening of fibrosis (49% and 39%, respectively, vs. 22%), improvement in fibrosis stage of at least 1 without worsening of NASH (48% and 34%, respectively, vs. 29%), and resolution of NASH plus improvement in fibrosis stage of at least 1 (35% and 25%, respectively, vs. 9%). Liver enzyme levels decreased and the levels of the majority of lipid, inflammatory, and fibrosis biomarkers improved in the lanifibranor groups. The dropout rate for adverse events was less than 5% and was similar across the trial groups. Diarrhea, nausea, peripheral edema, anemia, and weight gain occurred more frequently with lanifibranor than with placebo.
Conclusions
In this phase 2b trial involving patients with active NASH, the percentage of patients who had a decrease of at least 2 points in the SAF-A score without worsening of fibrosis was significantly higher with the 1200-mg dose of lanifibranor than with placebo. These findings support further assessment of lanifibranor in phase 3 trials. (Funded by Inventiva Pharma; NATIVE ClinicalTrials.gov number, NCT03008070. opens in new tab.)
| |
| |
| |
|
|
|