 |
|
|
| |
Summary from EASL 2013 for Hepatitis C - New HCV DAAs on their way soon: what do the phase III studies tell us?
|
| |
| |
Jürgen K. Rockstroh M.D., Professor of Medicine
University of Bonn, Germany
Correspondence:
Prof. Dr. J.K. Rockstroh
Department of Medicine I
University of Bonn
Sigmund-Freud-Str. 25
53105 Bonn
Germany
Introduction
Ever since the first DAA based triple therapy for HCV became available treatment paradigms for HCV therapy have subsequently changed significantly and promise cure of HCV infection in around two thirds of treatment naïve HCV-genotype-1 infected patients undergoing triple therapy. Nevertheless, with more widespread use of boceprevir and telaprevir based triple therapy the current challenges of HCV therapy have become quite evident: Adherence issues due to high pill burden and food requirements with tablet intake, high rate of adverse events particularly in patients with more advanced liver disease, multiple drug-drug interactions and finally also significantly lower response rates in more challenging patient populations (cirrhosis, previous null-response to dual therapy, post-transplant treatment etc.). So not so surprisingly after an initial euphoria to offer triple therapy to HCV patients who had waited a long time for more efficacious HCV treatment options, many physicians and patients likewise now seem to wait for easier to take and potentially better tolerated and at best even interferon free new treatment options in the near future. At this year's EASL in Amsterdam with over 9600 delegates the phase III study results for the two "second wave HCV protease inhibitors" faldaprevir and simeprevir each in combination with pegylated interferon (PEG-IFN) and ribavirin (RBV) for HCV genotype 1 patients were presented as well as the phase III findings for the polymerase inhibitor sofosbuvir again in combination with PEG-IFN/RBV for treatment of genotypes 1,4,5 and 6. In addition phase III results of the first interferon free combination of sofosbuvir with ribavirin for treatment of genotype 2 and 3 were presented. As these new drugs have been or are about to be filed for licensing it can be expected that at least for the US, approval of these new HCV drugs can be expected 2013/2014. Therefore, with most likely less than a year ahead before these new drugs hit the market the question who to treat now and in whom to wait for improved HCV treatment options has become even more pertinent.
But clearly there is an even more promising future behind these new HCV agents, suggesting interferon free regimens will become available even in more difficult to treat patient populations and for all genotypes in the upcoming years. Again several studies which were presented at EASL allowed a glimpse into this highly promising future. Nevertheless, issues around the potential cost of the HCV drugs to come as well as the question which prediction factors allow us to decide which patient needs how many DAAs and what kind of combination suits which type of patient remain unanswered to the very day. Indeed the high number of compounds and combinations makes it increasingly difficult to follow all studies. Also 100% sustained virological response rates in easy to treat naïve HCV patients with early fibrosis stages and an IL28b CC genotype as well as a 1b infection cannot be automatically transferred to more challenging patient populations. In summary, the HCV field is moving fast and new HCV compounds promising simpler treatment regimens with increased tolerability and shortened treatment durations can be expected soon. In addition first interferon-free treatment approach for genotype 2 and 3 will be available shortly. A successful interferon-free HCV treatment strategy for all patients however, still has not yet arrived.
Why treat hepatitis C?
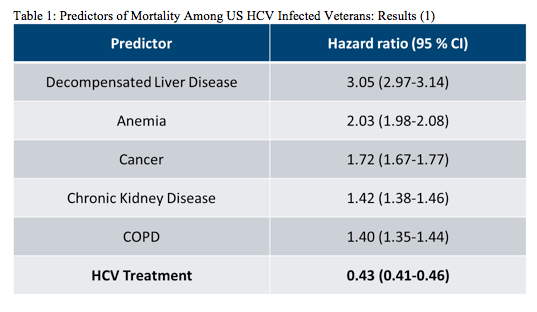
In consideration of the high costs of HCV therapy many countries in Europe have restricted the use of HCV protease inhibitor based triple therapy to more advanced fibrosis stages. Therefore, it is important that studies look at the impact of HCV therapy on survival in HCV infected individuals in order to demonstrate the benefits of HCV therapy and ultimately demonstrate cost-effectiveness of these treatment approaches. Interesting data in this context was presented at the EASL conference from a retrospective cohort study using the Electronically Retrieved Cohort of HCV Infected Veterans (ERCHIVES) (1). Within this study the predictors of mortality among US HCV infected veterans were evaluated. Overall they were able to compare an impressive number of 195,585 HCV Patients with 202,739 non-HCV infected veterans. The all-cause mortality rate among Veterans with HCV infection was much higher with 43.9 (43.4-44.3) per 1000 person-years (PY) than in Veterans without HCV infection (all-cause mortality was 24.0 (23.7-24.4) per 1000 PY). The table 1 shows the relevant predictors found to impact mortality. Among Veterans with HCV infection, decompensated liver disease, anemia, cancer, chronic kidney disease and COPD were the strongest predictors of higher risk of mortality, while HCV treatment was associated with over 50% reduction in mortality in this group. These findings once again underline the importance of HCV therapy to significantly lower risk of mortality in HCV infected individuals.
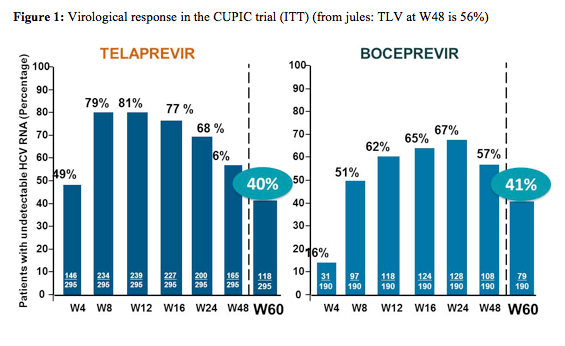
Efficacy and safety of boceprevir and telaprevir in clinical reality
The French early access program for boceprevir and telaprevir in cirrhotic previous interferon/ribavirin non-responders allowed studying efficacy and safety of the 2 new oral HCV protease inhibitors in a probably much more typical patient population seen in clinics today than in the more restricted clinical phase III trials (2). Only patients with compensated child A cirrhosis, HCV genotype 1 infection and prior relapse or partial response to PEG-IFN/RBV regimen were prospectively included to receive either 12 weeks of TVR, PEG-IFN-alfa2a/RBV and 36 weeks of PEG-IFN-alfa2a/RBV, or 4 weeks of PEG-IFN-2b/RBV and 44 weeks of BOC, PEG-IFN-2b/RBV without randomization which precludes any comparison between the two molecules (2). Overall 295 patients receiving telaprevir based HCV therapy and 190 receiving boceprevir-based HCV therapy were available for these analyses at week 60 resembling SVR 12 data. Most strikingly, despite very impressive early treatment responses with 81% of patients being undetectable at week 12 who were receiving telaprevir based triple therapy and 65% of patients being undetectable after 12 weeks of boceprevir containing triple therapy (so week 16 as patients started with a lead-in of 4 weeks dual therapy) SVR12 cure rates were substantially lower with 40% of patients being undetectable 12 weeks after discontinuing telaprevir based HCV therapy and quite similar with 41% being undetectable 12 weeks after stopping boceprevir based triple therapy. The rate of patients with an undetectable HCV-RNA for the two different HCV therapies for all relevant time points is shown in figure 1.
Further sub analyses revealed that the highest SVR12 rates were obtained in previous relapsers with 53% and 51% being undetectable at week 60 whereas lower response rates of only 29% and 11% were achieved in previous null-responders for telaprevir and boceprevir treated subjects, respectively. These findings once again emphasize that cirrhotic, previous null-responders remain a very challenging patient group to treat and that low cure rates need to be balanced against increased risk for adverse events. Patients with genotype 1b infection achieved SVR significantly higher than patients with genotype 1a infection (46 vs 34% in the telaprevir study and 51 vs 31% in the boceprevir study, respectively).
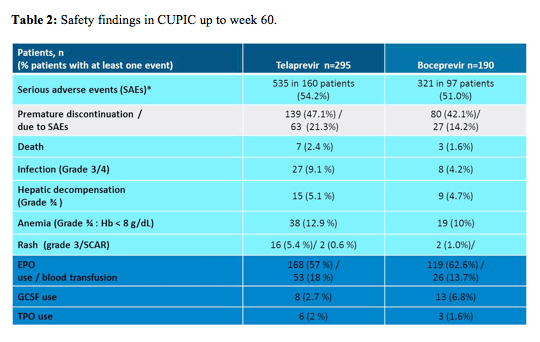
The main safety findings of the CUPIC trial are summarized in Table 2 and show that there is in particular an increased risk for infections as well as hepatic decompensation reminding us that the treatment of HCV infected patients with more advanced liver disease should preferably be conducted in specialized centers with special knowledge in the management of cirrhotic HCV patients. Highest risk of occurrence of death or severe complications was found in patients with low platelets < 100.000/mm³ and albumin < 35g/l (8% event rate). Several German groups also reported back their real world experience in administering HCV protease inhibitor based therapy in compensated cirrhosis. In one small study from Hannover in 48 patients the authors found an increased risk of treatment failure or development of a SAE in HCV patients with compensated cirrhosis and a platelet count <110/nl or a Child-Pugh Score >5 (3). In a study from Hamburg in 143 patients of whom 48% had advanced fibrosis, again platelets <100.000/μl and an albumin <40g/l were associated with treatment failure and SAE occurrence (4). Finally in an oral presentation at EASL, investigators from Austria presented their experience from treating cirrhotic patients and which predictive factors may exist for the emergence of septic events (5). In 131/191 patients with F3/F4 fibrosis triple therapy (65 telaprevir, 66 boceprevir) was associated with a poor outcome and a high rate of severe adverse events including three cases of death. Low platelet count, low serum albumin and HVPG ≥10mmHg were predictive markers for severe septic events. The authors suggested that these high risk patients may benefit from antibiotic prophylaxis on triple therapy.
Noteworthy in CUPIC is also the high rate of anaemia in both treatment arms as well as the high use of growth factors in particular erythropoietin in this study which highlights that anaemia management is of utmost importance in management of cirrhotic HCV patients under currently available triple HCV therapy. In a further sub analyses it was found that in particular age >65y , female gender, no lead-in and a baseline hemoglobin level of ≤12 g/dL for females and ≤13 g/dL for male were independently associated with a higher risk for developing severe anemia < 8g/dl or blood transfusion.
In conclusion it needs to be stressed that the safety profile of TVR or BOC with PEG-IFN/RBV in cirrhotic patients is poor and is associated with increased discontinuation rates due to SAEs compared to those reported in phase III trials. In particular patients with platelets below 110.000/μl and lower albumin levels < 35-40 g/l are at high risk of SAEs and should only be treated in very experienced centers. Cure rates are lower in the context of cirrhotic non-responders but are higher than 50% in previous relapsers. These data strongly suggest that triple therapy must be administered cautiously with intensive safety monitoring in patients with liver cirrhosis.
Telaprevir and boceprevir in clinical practice: what else can we learn?
One of the raised critical comments towards the CUPIC result is that physician's inexperience with the new HCV compounds may have also affected the safety management and partially may explain the somewhat higher SAE rate observed than in other cohorts. Taking into account the multiple inclusions and exclusion criteria for HCV studies these days cohort data becomes ever so important to get a feeling for efficacy and safety in a more "real world clinic" setting and to potentially address questions which can only be answered confidently with thousands of patients. Therefore, it was enlightening to see the first presentation from the TARGET cohort which has the aim to evaluate the effects of triple therapy in the broader population now being treated in the United States and Canada, including those underrepresented in clinical trials (6). The HCV-TARGET consortium of academic and community investigators utilizes novel, standardized source data abstraction and a common database to enroll sequential patients treated with regimens that include boceprevir and telaprevir.
Demographic, clinical, adverse event and virological data are collected throughout treatment and post-treatment follow-up. Whole blood for DNA and serum from specified time points are stored at a central bio repository. The current analysis includes available data as of April 2013. In this ongoing study, 1919 participants have been enrolled to date of whom 1457, at varying stages of treatment (telaprevir 1097, boceprevir 342), are included in this preliminary analysis. The majority of patients included are male (60%), Caucasian (72%), and have a mean age of 62 years. African Americans comprised 21% of participants and 7% of patients were older than 65 years. Cirrhosis, defined by biopsy or clinical criteria, was present in 38%. Genotype 1a/1b/not subtyped was 58%, 19%, 14%, respectively, and 49% were treatment naïve. So far 23% of patients on treatment discontinued therapy early mostly for virological failure (8% of the total patient population) or adverse events failure (9% of the total patient population). In this first interim analysis, the safety and on-treatment efficacy of telaprevir and boceprevir in the real world setting appeared to be comparable to that observed in registration trials. Although no new safety signals were observed, anemia emerged as the most relevant adverse event impacting clinical care. Ribavirin dose reductions were frequent and minimized the use of EPO and/or transfusions. Patients with cirrhosis in particular were at increased risk for severe anemia and early treatment discontinuation due to adverse events. Indeed hepatic decompensation events occurred in 11% of patients with cirrhosis and 2 of these died from sepsis-related events. So despite comparably high rates of hepatic decompensation in the more advanced liver disease patient fortunately very few patients were lost under therapy suggesting that careful monitoring and straight forward AE management can help to minimize therapy associated deaths. SVR rates are not yet available for the cohort but will become available soon. Predictors of adverse outcomes are also under investigation and will be reported soon as well.
EASL: SVR12 rates and safety of triple therapy including telaprevir or boceprevir in 485 cirrhotic non responders treated in the French Early Access Program (ANRS CO20-CUPIC) - (04/27/13)
Telaprevir and boceprevir: what else is new?
As it has become apparent that that there are patient groups which may respond particularly well to HCV DAA based triple therapy the question has emerged whether there may be patient groups which could be successfully treated with only 12 weeks of HCV therapy with telaprevir/PEG-IFN/RBV. Indeed exactly this question was studied in the CONCISE study (7). Within the CONCISE study treatment-naïve or prior relapse non-cirrhotic patients with IL28B CC genotype received T/PR for 12 weeks (T 1125mg bid, P 180mg/week, weight-based R 1000 or 1200 mg/day). Patients with Rapid Virologic Response (RVR; Week 4 HCVRNA <25 IU/mL, target not detected) and who continued all study drugs through the first 12 weeks of therapy were eligible for randomization (2:1) at week 12 to receive no further treatment (T12/PR12) or 12 additional weeks of PR (T12/PR24). Subjects without RVR or not completing 12 weeks of TPR were not randomized and were assigned to 24 or 48 weeks total therapy. SVR rates were assessed 4 (SVR4) or 12 (SVR12) weeks after end of planned treatment.
65.1% of patients had genotype 1a, the median HCV-RNA was 5,945,000 IU/ml. 86% of patients included were treatment naïve, median age was 47.4 years. Overall 238 subjects were included into the trial of whom 158 were randomized. 80 subjects were not randomized because of not completing the first 12 weeks of triple therapy (19 did not complete 12 weeks of drug but did achieve eRVR and 61 did not complete 12 weeks of drug and/or did not achieve eRVR). Of the patients randomized 2:1 106 were only treated for 12 weeks with telaprevir/PEG-IFN/RBV and 52 received a further tail of 12 weeks of PEG-IFN/RBV. High SVR 4 rates were found for both study arms with 100% of patients being undetectable in the 24 week treatment arm versus 89% in the 12 week treatment arm, respectively. SVR 12 rates remained high with 97% in the 24 week arm and 87% in the 12 week arm. Among all patients, the most commonly observed adverse events in the telaprevir treatment phase were fatigue (48%), nausea (47%), anemia (41%), headache (26%), pruritus (33%), pyrexia (22%) and rash (26%).
The authors concluded that high SVR rates from the CONCISE study analysis suggest the potential for the defined IL28B CC patients with rapid virological response to shorten duration of telaprevir plus peg-interferon/ribavirin to 12 weeks. The safety profile was considered to be similar to that seen in previous clinical trials with telaprevir combination treatment. This study suggests that markers such as IL28b may still play a role in the future as prediction factors which may allow shortening treatment durations of HCV therapy.
A further possibility to simplify telaprevir based therapy is to administer telaprevir bid which could be helpful in increasing adherence. Taking into account the necessity to take telaprevir together with a fat-containing meal a bid regimen may make the handling of the dietary requirements of a telaprevir based therapy much easier. Results from the OPTIMIZE study presented at AASLD in 2012 had previously established the non-inferior efficacy of telaprevir (TVR, T) twice daily (bid) versus every 8 hours (q8h) in combination with peginterferon/ribavirin (PR) across a range of patient characteristics. One stratification factor of the study was IL28B genotype. At the EASL conference the study group now presented the study results across IL28B genotype groups which are depicted in figure 2 (8).
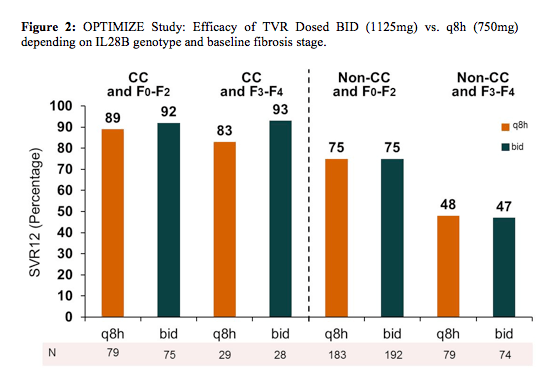
OPTIMIZE was a randomized, open-label, multicenter, Phase III trial in treatment-naïve patients with chronic HCV genotype 1 infection (NCT01241760). In total, 740 patients were stratified by IL28B genotype (CC, CT, TT) and liver fibrosis stage (F0-F2 vs. F3/F4), and randomized to either TVR 1125mg bid (N = 369) or TVR 750mg q8h (N = 371) in combination with PR. The percentage of patients by IL28B genotype was: CC=29%, CT=56%, TT=16%. The efficacy of TVR bid versus q8h was similar regardless of IL28B genotype (Figure2). SVR12 was higher in CC versus non-CC genotypes (90% vs. 67%, p < 0.0001). IL28B CC genotype was strongly associated with SVR12 after adjustment for other baseline factors, including fibrosis stage (OR = 5.00, 95% CI: 3.01, 8.30; p < 0.0001). When IL28B and fibrosis stage were considered together, the highest SVR12 (90%, 95% CI: 84%, 94%) was observed in CC patients with F0-F2 fibrosis stage, while the lowest SVR12 (47%, 95% CI: 39%, 56%) was observed in non-CC patients with advanced fibrosis or cirrhosis (F3-F4).
These findings clearly underline that the relative efficacy of TVR bid versus q8h is similar regardless of IL28B genotype baseline fibrosis stage and so all patient types can benefit from this treatment simplification.
More clinical response and safety data was presented in a late-breaker poster from a meta-analyses evaluating safety and efficacy of boceprevir based triple therapy for treatment of HCV genotype 1 patients with compensated cirrhosis (9).
As this patient group was somewhat underrepresented in some of the past trials this analyses allowed to look at larger patient numbers by including data from five phase II clinical trials. All analysed patients received 4 weeks of P/R lead-in followed by BOC/P/R or P/R for 24, 32 or 44 weeks. Metavir scores (centrally-read) of liver biopsies from 2415 patients showed: 2074 F0-F2; 129 F3; 212 F4. Multivariate logistic regression (MLR) models were used to identify baseline and on-treatment predictors of sustained virologic response (SVR), combining F3/F4 patients to increase power to identify predictors. The SVR percentage is shown below in figure 3:
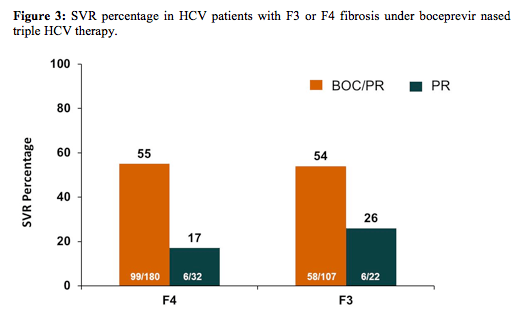
Multivariate logistic regression identified 4 predictors of SVR in F3/F4 patients: undetectable HCV-RNA at treatment week (TW) 8 (i.e., after 4 weeks of P/R lead-in + 4 weeks of BOC/P/R; odds ratio (OR) = 10.57; p < 0.0001); ≥1 log10 decline in HCV-RNA at TW4 (i.e., after 4 weeks of P/R lead-in; OR = 2.64; p = 0.0053); male sex (OR = 2.23; p = 0.0141) and baseline HCVRNA ≤800,000 IU/mL (OR = 2.55; p = 0.0383). No F3 (0/5) or F4 (0/17) patients with <3 log10 decline and detectable HCV-RNA at TW8 achieved SVR. Overall it appears that early time points during HCV therapy may be established to reliably predict outcome therapy possibly allowing treatment discontinuation in those with no chance of cure.
EASL: Efficacy of Telaprevir Dosed Twice Daily versus Every 8 Hours by IL28B Genotype: Results from the Phase III OPTIMIZE Study - (05/07/13)
EASL: High SVR Rates (SVR4) for 12-Week Total Telaprevir Combination Therapy in IL28B CC Treatment-Naïves and Prior Relapsers With G1 Chronic Hepatitis C: CONCISE Interim Analysis - (05/07/13)
Boecprevir or telaprevir in renal impairment
At this year EASL several studies addressed the use of DAAs in renally impaired patients. The prevalence of chronic hepatitis C (CHC) in Hemodialysis population is 3%. Clearly in the past treatment of HCV in renal impairment and especially patients on hemodialysis has been very difficult due to increased toxicity in these patients and the fact that ribavirin is cleared through the kidney and can mostly only be used in small doses. Indeed standard of care (SOC) offers reduced dose of Peg IFN Alfa (p-IFNa) and reduced Ribavirin doses eliciting sub optimal SVR of only 27%. Also in HCV positive patients receiving kidney transplant interferon based therapy has been associated with higher organ rejection rates so getting rid of HCV is important for subsequent transplant outcome. Telaprevir or boceprevir are not excreted renally and insignificant amount of the drugs is removed through hemodialysis therefore treatment with these drugs appears feasible. One poster from the US looked at the safety of HCV PI based triple therapy in 14 HCV genotype 1 patients with end stage renal disease (ESRD), of which 8 were being evaluated for kidney- and 6 for combined liver kidney transplantation (10). All patients received pegylated interferon alpha 2a (P), ribavirin (R) and either boceprevir (B) 800mg thrice daily or telaprevir (T) 750mg thrice daily. All patients treated with BPR received 1 month lead-in, P 180 micrograms weekly and R 200mg thrice weekly whereas those treated with TPR received no lead-in, P 135micrograms weekly and R 200mg - daily (12.5%), twice weekly (50%) and thrice weekly (37.5%) at the discretion of the clinician.
Patients included into the analysis showed a mean age of 56 years, 71% were male, 57% African-Americans, 57% had genotype 1a, 71% had advanced fibrosis and 100% of patients were characterized by the unfavourable IL28b non CC genotype. Mean baseline haemoglobin was 12.3 g/dl. Overall, 8 hospitalizations occurred in 5 patients (4 due to anemia, 2 due to pneumonia/infection, 1 ascites and 1 due to dehydration). 85% of patients so far completed 12 weeks of therapy. Anemia occurred in 50% in both groups but more aggressive management was required in the TPR group. Indeed all telaprevir treated patients had at least a drop in haemoglobin of 1g/dl and 75% a drop >2g/dl versus only 67% and 60% of the boceprevir treated patients having a >1 or >2g/dl frop in haemoglobin, respectively.
In summary, adverse event rate was high but triple therapy appeared feasible under close monitoring. SVR rates of treatment are still pending.
In the oral abstract session (11) another study evaluated the triple therapy in naïve CHC-G1 in hemodialysis in Respond Guided Therapy (RGT). Overall a total of 36 treatment-naïve genotype 1 patients were recruited. 26 patients were male 10 female. Mean age was 58 years, mean BMI 26.6%, 41% had genotype 1a, 64% were black and 17% Hispanic. Only 27% of patients had the favourable IL28B CC genotype and most patients had advanced fibrosis (72% F3, 14% F4). Mean haemoglobin was 11.8 g/dl, all patients received EPO during hemodialysis per weekly schedule. Patients were randomized into 3 groups: Group A (n = 12): p-IFNa 135mcg once weekly, Telaprevir 750mg two tablets-TID four days and three tablets BID post dialysis for three days; along with RBV 200mg daily for 12 weeks followed by p-IFNa 135mcg plus RVB 400mg till 24 weeks. Group B (n = 12) p-IFNa 135 mcg once weekly with Telaprevir same as Group A with Placebo for 12 weeks followed by p-IFNa 135mcg with RBV 400mg till 36 weeks. Group C (n=12) received p-IFNa 135mcg once weekly, Placebo and RBV 400mg daily for 48 weeks. SVR24 rates were highest in group A (week 48) with 63%, followed by group B with 50% (week 60) and lowest in group C with dual therapy of only 25% (week 72). So clearly this study suggests that the addition of the HCV protease inhibitor substantially adds to HCV cure rates. In group A despite shortened treatment duration of 24 weeks only one relapse was noted versus none in group B and one breakthrough in group C. Safety wise anemia was clearly more pronounced in the telaprevir arms with anemia rates of 58% in group A and 50% in group B versus 33% in the dual therapy arm. These results are very encouraging and definitely warrant an evaluation in a larger study.
Phase III studies with new DAAs in clinical development in combination with pegylated interferon and ribavirin
New HCV protease inhibitors in combination with PEG-IFN/RBV
At this year EASL an astounding number of phase III final results were presented, offering safety and efficacy data of new compounds in larger patient sets than studied before and thereby providing a glimpse on the performance of drugs which most likely will become available in the very near future in 2014. The first three phase III studies were around the combination of the new second wave HCV protease inhibitors faldaprevir and simeprevir.
The STARTVerso1 was a double-blind, placebo-controlled Phase III study which assessed the efficacy and safety of faldaprevir (FDV) plus pegylated interferon alfa-2a and ribavirin (PegIFN/RBV) (12). Overall 652 treatment-naïve HCV genotype 1 patients were enrolled into the study and randomised 1:2:2 to receive 24 weeks PegIFN/RBV with placebo for 24 weeks (arm 1); FDV 120mg QD for 12 or 24 weeks (response guided; arm 2); or FDV 240mg QD for 12 weeks (arm 3). Patients with early treatment success (ETS, HCVRNA < 25 IU/mL at week 4 and undetectable at week 8) in arms 2 and 3 stopped all treatment at Week 24. Patients without ETS and those in arm 1 continued to receive PegIFN/RBV for 48 weeks.
Randomisation was stratified by HCV GT-1 subtype and race. The primary endpoint was sustained virological response 12 weeks after planned end of treatment (SVR12). The study design is depicted below in figure 4.
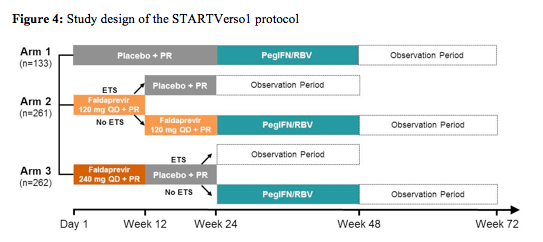
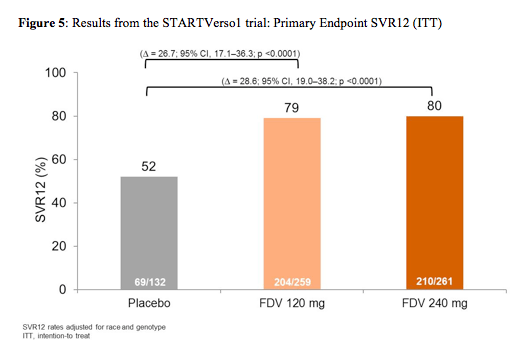
652 patients were treated: mean age 48 years, 52% male, 78% Caucasian, 20% Asian, 17% grade ≥3 fibrosis, 39% IL28B CC, 66% GT-1b. Noteworthy, is that most likely due to the fact that parts of the study were partially done in Japan a much higher rate of genotype 1b was found at baseline compared to many other DAA trials as well as a higher rate of the more favourable IL28b CC genotype. This may also explain the relative high rate of SVR12 in the dual treatment arm with PEG-IFN/RBV. Nevertheless, the addition of faldaprevir was associated with a drastic increase in SVR12 rates (see figure 5) allowing for cure of HCV in 79-80% of patients. No difference in response rate was found for the two doses tested, respectively. Early treatment success (ETS) defined as HCV RNA < 25 IU/mL (detected or undetected) at Week 4 and < 25 IU/mL (undetected) at Week 8 which allowed to go for 24 weeks of treatment only was achieved in 87% of the 120mg QD arm and 89% in the 240mg QD arm with 86% and 89% achieving ETS and SVR12, respectively in the two dose arms. Clearly this means that for the majority of treatment naïve GT1 patients treated with this combination shortened treatment durations of 24 weeks are feasible. Comparing GT 1a and 1b patients it becomes evident that GT 1a patients are more difficult to treat with only 36% of patients receiving dual therapy in the control arm achieving SVR12 versus 69% in the 120mg QD arm and 76% in the 240mg QD arm. In contrast higher response rates of 60% in the control arm, 84% in the 120mg QD arm and 83% in the 240mg QD arm were documented for GT1b patients, respectively.
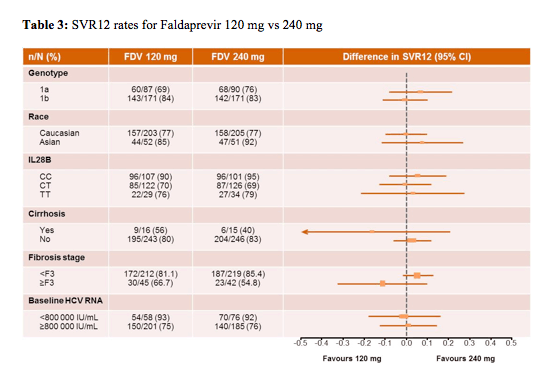
When comparing the different doses of faldaprevir studied with respect to various baseline parameters no significant difference was found between the two doses regardless of baseline fibrosis level, IL28b genotype, race, HCV 1 subtype or baseline HCV-viremia (see table 3).
Looking at the safety results all study medications were discontinued due to adverse events (AEs) in only 4% in the control group, 4% in the 120mg QD group and 5% of patients in the 240mg QD group. FDV only was discontinued in 0%, 1% and 3% of patients, respectively. Serious AEs occurred in 6%, 7% and 7% of patients. Grade 3 rash occurred in <1% of patients in each arm; no patients had Grade 4 rash. Up to Week 24, haemoglobin ≤8.5 g/dL occurred in 2%, 3% and 3% of patients, respectively.
In conclusion, FDV plus PegIFN/RBV significantly increased SVR12 rates in HCV GT-1 patients in Europe and Japan compared with PegIFN/RBV alone and was well tolerated. In total, 88% of patients treated with FDV were eligible to stop all treatment at Week 24. These are overall excellent results for a mostly shortened treatment duration but are clearly impacted by the high proportion of HCV GT1b patients included into the study.
The next second wave HCV protease inhibitor evaluated was simeprevir formerly known as TMC-435 which was examined in two similar studies named Quest 1 and 2 (13,14). QUEST-1 (TMC435-C208; NCT01289782) and QUEST-2 (NCT01290679) were both Phase III, randomised, double-blind, placebo controlled trials assessing the efficacy, safety and tolerability of simeprevir versus placebo as part of a regimen including peginterferon/ribavirin (PR) in treatment-naïve patients chronically infected with genotype-1 HCV. HCV genotype-1-infected patients with METAVIR score F0-F4 (QUEST-1 n = 394; QUEST-2 n=391), stratified by HCV subtype and host IL28B genotype, were randomised 2:1 to receive simeprevir (150mg QD) or placebo, plus PR for 12 weeks, followed by PR alone. In QUEST-1 all patients received peginterferon a-2a 180μg/week + ribavirin 1000-1200mg/day. In QUEST-2 63% of patients were randomised to receive SMV or placebo + PegIFNα-2a or 2b/RBV; the remainder received PegIFNα-2a/RBV. Total treatment duration was 24 or 48 weeks (simeprevir group) based on response-guided therapy (RGT) criteria (HCVRNA< 25 IU/mL Week 4 and undetectable Week 12) or 48 weeks (placebo group). The study design is shown below in figure 6.
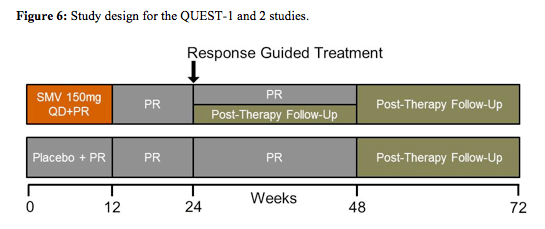
Stopping rules were as follows: if HCV RNA > 1000 IU/mL week 4 stop SMV/placebo;
if HCV RNA <2log10 IU/mL reduction at Week 12, or confirmed
>25 IU/mL at Week 24 or 36, stop all treatment. In QUEST-1 baseline demographics showed 56% of patients in the simeprevir 150mg QD arm to have GT 1a infection vs. 57% in the control group. 12 and 13 patients had METAVIR score F4, respectively. 10% vs. 3% of patients were black/African American. In Quest-2 40.9% of the patients in the simeprevir arm had GT1a infection vs 40.3% in the control arm with dual therapy. 92.8 vs. 91.8% patients were white, 6.9 vs. 11% had METAVIR F4 fibrosis and 29.3 vs 31.3% had an IL28B CC genotype, respectively. Again this was a trial with a rather high inclusion of more favourable to treat HCV GT1b patients. The main results for the two studies are shown in figure 7.
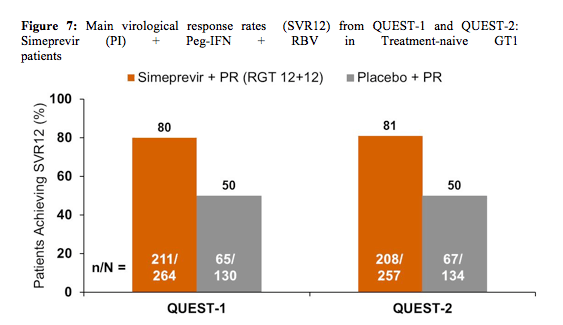
Overall, 85-91% qualified for shortened therapy. Response rates were impressive and clearly the addition of simeprevir significantly improved treatment response over dual therapy in all key subgroups including baseline HCV viral load, fibrosis stage, IL28b genotype and gender. In QUEST-2 statistically significantly higher SVR12 rates with SMV/PR compared with placebo/PR were also found irrespective of the type of PegIFN used. Interestingly though there was a trend for lower SVR12 rates for patients in the placebo arm receiving PegIFNα-2b/RBV. In QUEST-2 84.6% of patients with F0-2 fibrosis achieved SVR12 versus 64.7% in patients with F4 fibrosis. Despite low F4 patient numbers it looks as if more advanced stages of fibrosis perform poorer than earlier fibrosis stages.
Safety results overall were quite favourable. AEs led to treatment discontinuation in QUEST-1 in only 3% of simeprevir treated patients in QUEST-1 and in 1.6% of simeprevir treated patients in QUEST-2. In the simeprevir groups, transient and mild bilirubin elevations, not accompanied by increases in ALT, AST or alkaline phosphatase, were observed. The mechanism is attributed to inhibition of OATP1B1/MRP2 transporters.
In summary, SMV 150 mg QD + PR in HCV genotype 1-infected, treatment-naïve patients leads to significantly increased SVR12 rates compared with PEG-IFN/RBV alone in around 80-81% of patients. Overall, 85-91% qualified for shortened therapy of 24 weeks. Safety and tolerability was comparable to placebo.
Overall, the phase III studies for faldaprevir and simeprevir show significantly enhanced cure rates over dual therapy. In addition they offer simplified once daily administration and considerably improved tolerability and safety profiles. However, in patients with cirrhosis SVR rates seem to be considerably lower than in earlier fibrosis stages suggesting that more potent strategies will be needed to cure these particular patients.
EASL: FALDAPREVIR PLUS PEGYLATED INTERFERON ALFA-2A AND RIBAVIRIN IN CHRONIC HCV GENOTYPE-1 TREATMENT-NAIVE PATIENTS - (04/27/13)
EASL: Simeprevir (TMC435) with peginterferon/ribavirin for chronic hCV genotype 1 infection in treatment-naïve patients: results from QUEST-1, a Phase III trial - (04/25/13)
EASL: Simeprevir (TMC435) with peginterferon-α2a or -α2b and ribavirin in treatment-naïve HCV genotype 1 patients: QUEST-2, a randomised Phase III trial - (04/27/13)
Sofosbuvir in combination with PEG-IFN/RBV
Next to the new HCV protease inhibitors, phase III results were also presented for the HCV-specific nucleotide polymerase inhibitor (chain terminator) sofosbuvir in treatment-naive genotype 1,4,5 and 6 patients (15). These are therefore the first phase III data for other genotypes than genotype 1. In the NEUTRINO trial open label sofosbuvir (SOF) 400 mg QD + Peg-IFN-alfa-2a 180 μg/week + RBV 1000-1200 mg/day was studied for 12 weeks (no response-guided therapy arm!). The study aimed at enrolling a target of 20% patients with cirrhosis. The broad inclusion criteria put no upper limit to age or BMI and permitted opioid replacement therapy. The study design is shown below in figure 8.
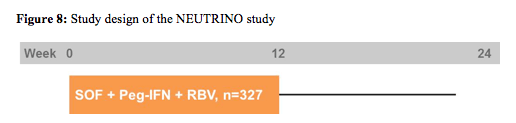
SVR12 was the primary efficacy endpoint defined as HCV RNA < LLOQ at post-treatment Week 12. HCV RNA was analysed by COBAS® TaqMan® HCV Test v2.0 HPS, with LLOQ of 25 IU/mL. A prespecified comparison to a historical SVR control rate of 60% for PEG-IFN/RBV was planned. Overall, 327 patients were included. The baseline demographics were as follows: 17% black, 89% GT1, 17% cirrhosis and 29% the favourable IL28b CC genotype. The main results are depicted below in figure 9.
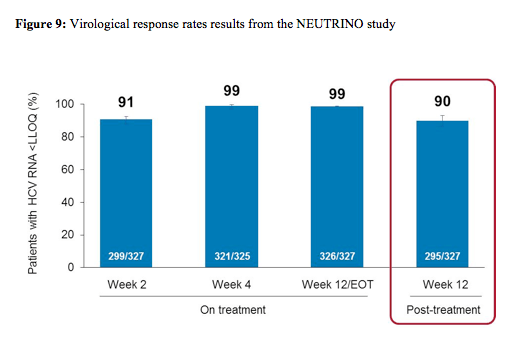
An impressive 90% of patients achieved SVR12 after only 12 weeks of sofosbuvir + PEG-IFN/ribavirin. Thereby, the study met the primary endpoint of superiority over historic control rate of 60% (p <0.001). Relapse accounted for all virologic failures. No resistance (deep sequencing) in any relapse patient was found. 89% of GT1 patients versus 96% of GT4 patients and 100% of GT 5,6 patients achieved SVR12. Virologic response by cirrhosis
status revealed slightly lower cure rates in patients with cirrhosis (80 vs 92%) versus non-cirrhotic patients. The safety profile was very favourable with a low rate of SAEs (only 4 patients) and only 5 treatment discontinuations because of AEs.
In summary SOF + Peg-IFN + RBV for 12 weeks resulted in an impressive 90% SVR12 rate, thereby being statistically superior to historical control SVR rate of 60%. Genotype 4, 5, and 6 HCV-infected patients had a 97% SVR12 rate demonstrating good activity in other than GT 1 patients. SOF was well tolerated without any additive effects of SOF to the expected safety profile of Peg-IFN + RBV
Phase III trials for HCV genotype 2 and 3
At this year EASL next to 3 phase III trials for GT1 patients also 3 phase III results for treatment of HCV genotype 2 or 3 infection were presented. The studies were conducted in treatment-naïve, treatment experienced and patients ineligible/intolerant to PEG-IFN/RBV.
Within the FISSION trial patients infected with HCV genotype 2 or 3 were randomized to sofosbuvir (400mg once daily) and ribavirin or PEG-IFN/RBV. RBV dose was 1000-1200 mg/day for SOF + RBV and 800 mg/day for Peg-IFN + RBV (16). Figure 10 below shows the corresponding study design. The primary endpoint was SVR12 with a pre-specified non-inferiority margin of 15%.
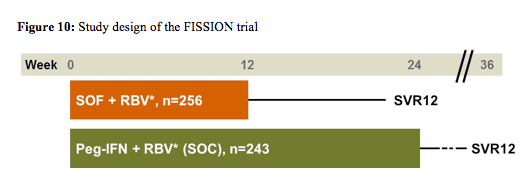
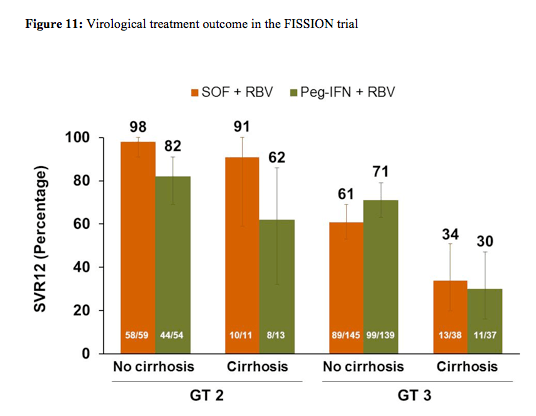
Baseline demographics were well balanced between the study arms. 87% of patients from both arms were white, 43% in the SOF arm and 44% in the control arm with dual therapy had an IL28b CC genotype, and 20% vs. 21% had cirrhosis at baseline. Overall, 72% of patients in both arms had a genotype 3. The main virological outcomes are shown below in figure 11. Overall, 67% of patients achieved SVR12, therefore the endpoint of non-inferiority for sofosbuvir + ribavirin versus PEG-IFN/RBV was reached. Overall, 97% of the HCV genotype 2 patients responded successfully to sofosbuvir + ribavirin versus only 56% in the HCV genotype 3 patients. Excellent HCV cure rates were observed for non-cirrhotic GT2 patients in both treatment arms, which seemed to be maintained even in cirrhotic GT2 patients with the IFN-free sofosbuvir/ribavirin combination, whereas cure rates for PEG-IFN/RBV dropped in the cirrhotic patient group. Genotype 3 patients were obviously more difficult to treat particularly in patients with cirrhosis with only 34% reaching SVR12 under the sofosbuvir combination and 30% under dual therapy in the control arm. Overall relapse rates were 30% in the sofosbuvir arm and 21% in the PEG-IFN/RBV arm.
None of the discontinuations due to AEs in the SOF+RBV arm was deemed related to SOF. Initiation of treatment for depression was less common with SOF+RBV (2%) versus PEG+RBV (11%). Discontinuation of treatment because of AEs occurred in 1% of sofosbuvir treated patient versus 11% of PEG-IFN/RBV treated patients.
In conclusion, SOF+RBV demonstrated non-inferiority to PEG-IFN+RGV. Overall, GT2 patients responded extremely well with 97% cure rate under the IFN-free strategy. For genotype 3 patients however the low response rate in particular in cirrhosis patients suggests that either more potent treatment strategies will be needed (for example adding a further DAA) or a possibly a longer treatment duration. SOF+RBV for 12 weeks were well tolerated in treatment-naïve cirrhotic and non-cirrhotic GT2/3 HCV-infected patients. Fewer AEs, discontinuations due to AEs, and Grade ¾ laboratory abnormalities were observed during treatment with SOF+RBV as compared to treatment with PEG+RBV.
The POSITRON study was a randomized (3:1), double blind, placebo controlled phase III study evaluating efficacy and safety of sofosbuvir + ribavirin in interferon ineligible/intolerant or unwilling HCV genotype 2 or 3 patients (17). Patients randomized to placebo were offered open-label SOF+RBV following study completion. The primary endpoint was SVR12. The figure 12 below shows the corresponding study design.
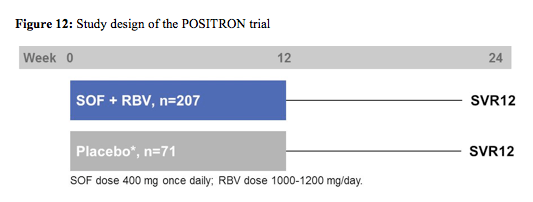
278 patients (44% IFN-ineligible, 9% IFN-intolerant, 47% IFN-unwilling) were randomized and treated; 54% were male, 91% white, 16% had compensated cirrhosis, 51% GT2, 45% were IL28B CC genotype. The SVR12 rate for SOF+RBV (161/207, 78%) was superior to placebo (0%, p < 0.001). All patients became HCVRNA negative on treatment, and relapse accounted for all virologic failures. SVR12 rates were 93% in GT2, 61% in GT3, 81% in non-cirrhotics, and 61% in cirrhotics. The difference in SVR12 by cirrhosis status was observed in GT3 patients only. The virological response rates are shown for the overall study outcome in figure 13 and with regard to fibrosis stage at baseline in figure 14.
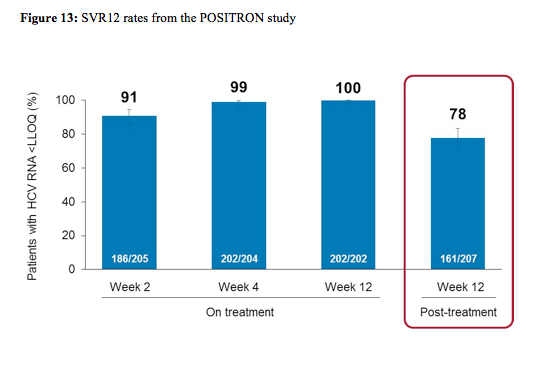
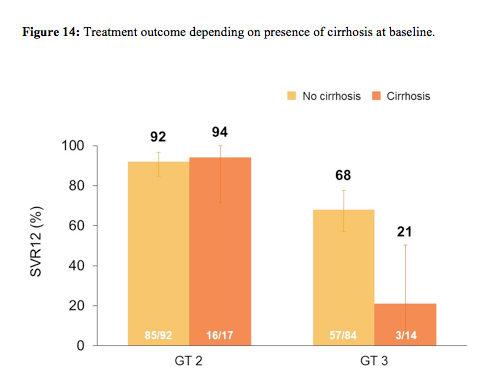
As in the FISSION trial treatment response was excellent for genotype 2 patients regardless of fibrosis stage. In genotype 3 patients however, only very low response rates were seen in cirrhotic patients again highlighting that different treatment strategies will be needed for this particular patient group. Sequencing performed on samples from 40/42 patients with relapse showed no S282T mutations by population or deep sequencing (1% cut-off). Safety again was very good with more treatment discontinuations because of AEs in the placebo group than in the sofosbuvir+ribavirin group (4% of patient's vs. 2%).
In summary, SOF+RBV for 12 weeks achieved a high SVR12 rate without evidence of resistance and was well tolerated in cirrhotic and non-cirrhotic GT2 and non-cirrhotic genotype 3 HCV-infected patients who do not currently have any treatment options. For cirrhotic genotype 3 patients more potent treatments need to be explored.
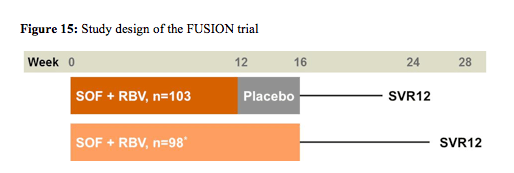
The last phase III trial reported the results from the FUSION trial which was a study evaluating safety and efficacy of sofosbuvir (SOF) 400mg once daily + ribavirin (RBV) 1000-1200mg in a divided daily dose for 12 or 16 weeks in treatment-experienced GT2/3 HCV-infected patients (NCT01604850) (18). FUSION was a placebo-controlled, double-blind study of treatment-experienced GT2/3 HCV-infected patients who were randomized 1:1 to SOF+RBV for 12 weeks followed by SOF placebo + RBV placebo for 4 weeks or to 16 weeks SOF+RBV (stratified by cirrhosis and HCV genotype). The primary endpoint was SVR12. The study design is shown below in figure 15.
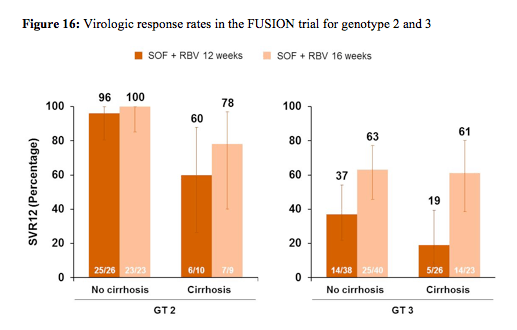
201 patients were randomized and treated; 70% were male, 87% were white, the mean age was 54 years (range 24-70), 30% were IL28B CC genotype, 34% had compensated cirrhosis, 63% had GT3 infection, 75% had relapse/breakthrough after prior treatment. No patients experienced on-treatment virologic failure (100% end-of-treatment response for both treatment arms). Relapse after EOT accounted for all virological failures and occurred in 50% of patients in the 12 week arm and in 27% of patients in the 16 week treatment arm, respectively. Most strikingly longer treatment duration of 16 weeks led to higher response rates in cirrhotic GT2 patients as well as in all GT 3 patients suggesting that longer treatment durations may help to improve the overall cure rates in GT3 patients. The virological responses rates for GT2 and 3 in relation to baseline cirrhosis are shown below in figure 16.
Sequencing was performed from samples of all 73 patients with relapse, but no S282T mutation was observed by population or deep sequencing (1% cut-off). One subject (<1%) discontinued treatment due to an AE. Eight subjects (4%) reported treatment-emergent SAEs.
In summary, in the FUSION trial in treatment experienced patients retreatment with sofosbuvir + ribavirin led to high SVR rates in non-cirrhotic HCV genotype 2 infected patients following treatment for 12 or 16 weeks. In the cirrhotic GT2 patients longer treatment of 16 weeks led to a numerically higher cure rate. Extending treatment duration to 16 weeks significantly increased SVR rates in genotype 3 HCV-infected patients, particularly those with cirrhosis.
Gilead Reports Interim Data From Phase 2 LONESTAR Study - (05/03/13)
EASL: Treatment With Sofosbuvir + Peginterferon + Ribavirin for 12 Weeks Achieves 90% SVR12 in Treatment-Naïve Genotype 1, 4, 5, and 6 HCV-Infected Patients: The NEUTRINO Study - (04/27/13)
EASL: Treatment With Sofosbuvir + Ribavirin for 12 Weeks Achieves SVR12 of 78% in GT 2/3 Interferon-Ineligible, -Intolerant, or -Unwilling Patients: Results of the Phase 3 POSITRON Trial - (04/27/13)
EASL: Phase 3 Randomized Controlled Trial of All-Oral Treatment With Sofosbuvir + Ribavirin for 12 Weeks Compared to 24 Weeks of PEG + Ribavirin in Treatment-Naïve GT 2/3 HCV-Infected Patients (FISSION) - (04/25/13)
EASL: All Oral Therapy With Sofosbuvir + Ribavirin for 12 or 16 Weeks in Treatment-Experienced Genotype 2/3 HCV-Infected Patients: Results of the Phase 3 FUSION Trial - (04/25/13)
Anything else for the treatment of HCV genotype 2 or 3 infection?
At EASL results from the COMMAND GT2/3 study were presented (18). Within this study the efficacy and safety of daclatasvir (DCV) + peg-alfa/RBV administered for either 12 or 16 weeks were compared with a standard 24-week course of peg-alfa/RBV in HCV GT 2 or 3 infection. The peg-alfa formulation used in this study was peg-alfa-2a (Pegasys®), ribavirin was dosed with 400 mg twice daily and daclatasvir was dosed with 60 mg once daily. The corresponding study design is depicted in figure 17.
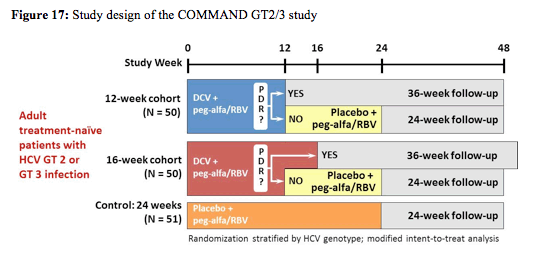
Adult treatment-naïve patients were randomly assigned to DCV/peg-alfa-2a/RBV for 12 or 16 weeks or placebo/peg-alfa-2a/RBV for 24 weeks. DCV/peg-alfa-2a/RBV recipients without protocol-defined response (PDR; HCVRNA < LLOQ week-4 and undetectable week-10) discontinued DCV at week 12 and received 12 additional weeks of peg-alfa-2a/RBV. The primary efficacy endpoint was SVR24. Baseline characteristics were well-balanced in the DC 12-week (N = 50), DCV 16-week (N = 50) and placebo (N = 51) arms; more patients with GT3 (18/80, 22.5%) than GT2 (1/71, 1.4%) were cirrhotic. Figure 18 shows the results separately for GT 2 and 3.
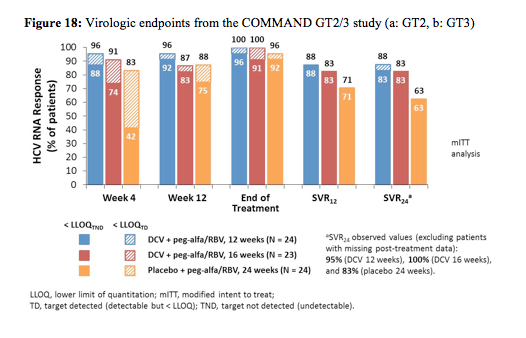
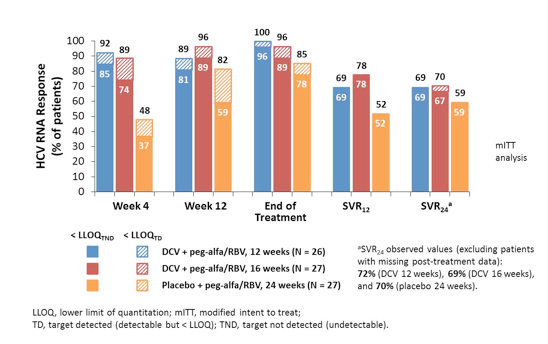
DCV + peg-alfa-2a/RBV treatment for 12 or 16 weeks achieved numerically higher SVR24 rates than 24 weeks of peg-alfa-2a/RBV in patients with GT 2 or GT 3 infection. Overall, however, SVR rates were higher in GT2 than in GT3, mostly because of higher relapse rates in GT3 again suggesting that perhaps different treatment algorithms are needed for GT2 infection than for GT 3 infection. No difference in response rates for GT3 was seen for 12 weeks versus 16 weeks of daclatasvir. Cirrhosis and resistance associated polymorphisms at baseline were found to be associated with an increased risk for relapse.
DCV + peg-alfa/RBV was generally well tolerated, with comparable spectra and frequencies of safety-related events compared with peg-alfa/RBV alone. Overall, 6% of patients in the daclatasvir 12 week arm versus 2% of patients in the daclatasvir 16 week arm versus 4% in the control arm with dual therapy discontinued treatment because of adverse events.
In summary, daclatasvir in combination with PEG-IFN/RBV achieved higher cure rates than PEG-IFN/RBV alone and allowed shortening treatment in the majority of patients. Nevertheless, treatment response rates in GT 3 patients are again lower as in GT2 particularly in cirrhosis.
EASL: DACLATASVIR COMBINED WITH PEGINTERFERON ALFA 2A ALFA-AND RIBAVIRIN FOR 12 OR 16 WEEKS IN PATIENTS WITH HEPATITIS C VIRUS GENOTYPE 2 OR 3 INFECTION: COMMAND GT 2/3 STUDY - (04/27/13)
Any news on interferon-free regimens for HCV genotype 1?
Another highlight of this year's EASL were clearly the high number of interferon-free treatment approaches which were presented or updated at the conference. After previous reports at other conferences that the IFN-free combination of sofosbuvir and ribavirin may achieve high cure rates in treatment naïve HCV genotype 1 patients but not in previous null responders great interest was directed towards new results from the ELECTRON study which attempted to study whether the addition of another DAA with a different mechanism of action and non-overlapping resistance profile would improve rates of SVR. Therefore in the ELECTRON study sofosbuvir (SOF) was administered in combination with GS-5885 (NS5A inhibitor; ledipasvir) plus RBV and GS-9669 (non-nucleotide NS5B inhibitor) plus RBV.
Non-cirrhotic patients with HCV genotype 1 were enrolled to receive 12-week regimens:
1. 25 treatment-naïve patients received SOF+ ledipasvir + RBV, 2. 9 prior null responders received SOF + ledipasvir + RBV, 3. 25 treatment-naïve patients received SOF + GS-9669+ RBV, and 4. 10 prior null responders received SOF+ GS-966 9+ RBV. Baseline demographics for the different patient groups showed that 80-90% of the included patients had GT1a HCV infection, with 0-20% of the previous null-responders having a favorable IL28b CC genotype versus 28-44% in the treatment-naïve patient groups. The virological response rates for the different treatment arms (so far available) are shown below in figure 19.
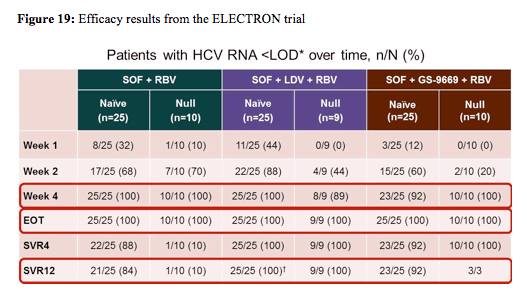
84% of treatment-naïve patients without cirrhosis achieved SVR12 after 12 weeks of SOF + RBV, but only 10% of prior null responders did so. Adding ledipasvir increased efficacy of SOF + RBV for 12 weeks substantially with 100% SVR12 in both treatment-naïve patients and prior null responders indicating that combination of DAAs may be a very successful strategy to improve response rates in more difficult to treat patient populations. No additional safety or tolerability issues were observed under the addition of ledipasvir. Adding GS-9669 also increased efficacy of SOF + RBV for 12 weeks with a 92% SVR12 in treatment-naïve patients. A relapse within 4 weeks of stopping therapy was recorded in 2 patients. Treatment outcome results for prior null responders are not yet available for all patients limiting further conclusions at this time point. In the patients who have already achieved the week 4 after stopping therapy the SVR4 rate in prior null responders so far is 100%. No additional safety or tolerability issues were recorded with the addition of GS-9669.
EASL: ELECTRON: All-Oral Sofosbuvir-Based 12-Week Regimens for the Treatment of Chronic HCV GT 1 Infection - (04/27/13)
Another much talked about presentation were the results from the AVIATOR study, which was a study that assessed safety and efficacy of regimens of ABT-450/r (HCV protease inhibitor dosed with ritonavir 100 mg, identified as a lead compound by Abbott and Enanta) with ABT-267 (NS5A inhibitor) and/or ABT-333 (non-nucleoside NS5B inhibitor) +/± ribavirin (RBV). Non-cirrhotic, GT1 treatment-naïve patients and prior peginterferon/RBV null responders received ABT-450/r (100/100-200/100mg QD) with 1-2 other DAAs (ABT-267 25mg QD, ABT-333 400mg BID) ± RBV for 8, 12, or 24 weeks. The corresponding study arms and the main virological outcome findings are shown below in figure 20.
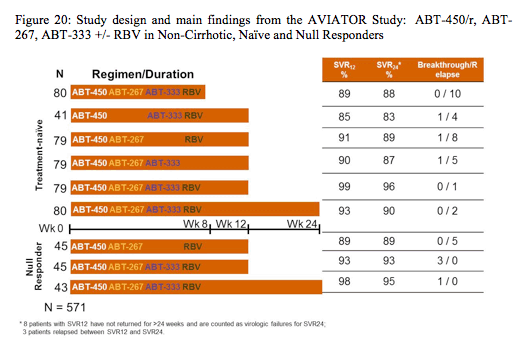
Overall 571 patients were included into this multiple arm study. A duration of 8 weeks with the 3 DAAs + ribavirin was associated with the highest number of relapses suggesting 8 weeks may be too short for some patients. Comparable responses were seen with 12 and
24 weeks of treatment, supporting selection of the 12-week duration of therapy in these populations. Among 247 subjects treated in the 12- and 24-week 3-DAA+RBV arms, 81.0% had IL28B non-CC genotype, 66.0% had GT1a infection, 54.8% were male and 10.5% were Black, with mean age of 51 years. Baseline HCV-RNA was 6.6 log10 IU/mL and 37.4% had ≥F2 fibrosis. SVR4 rates were comparable in the 12 and 24 week arms (98.7% and 96.2% in treatment naïve and 93.3% and 97.7% in prior null responders, respectively). SVR4 rates (combined 12 and 24 week arms) were comparable in patients with IL28B CC vs. non-CC, GT1a vs. GT1b, baseline HCVRNA <7 log10 vs. ≥7 log10 IU/mL, and F0-F1 vs. ≥F2 fibrosis.
Among these 247 subjects, 4 patients (1.6%) discontinued due to study drug-related AEs. Seven patients (2.8%) experienced SAEs; 1 (arthralgia) was possibly study drug-related. The moderate-to-severe study drug-related AEs with >10% incidence in any arm were asthenia and fatigue. 6 subjects (2.8%) and 1 subject (0.6%) experienced G3-4 laboratory abnormalities in total bilirubin and ALT, respectively; all resolved with continued dosing.
In conclusion in the AVIATOR study SVR rates >90% were achieved in naïve and prior null responder patients with a 3-DAA+RBV regimen, across HCV subtype, IL28B genotype, baseline HCV-RNA or severity of fibrosis. The 12-week duration of therapy in treatment-naïve and previous null-responders seemed to be the best duration choice in the studied populations.
EASL: SAFETY AND EFFICACY OF INTERFERON-FREE REGIMENS OF ABT-450/r, ABT-267, ABT-333 +/- RIBAVIRIN IN PATIENTS WITH CHRONIC HCV GT1 INFECTION: RESULTS FROM THE AVIATOR STUDY - (04/25/13)
Finally also an ongoing phase II study was presented which is evaluating the IFN- and RBV-free regimen of daclatasvir (DCV) (NS5A inhibitor), asunaprevir (ASV) (protease inhibitor) and BMS-791325 in two doses (nonnucleoside NS5B inhibitor, 75mg vs. 150mg BID) for 24 or 12 weeks. Treatment-naive, HCV GT1, non-cirrhotic patients (N = 32) were randomized 1:1 to DCV 60mg QD, ASV 200mg BID, and BMS-791325 75mg BID for 24 (Group 1) or 12 (Group 2) weeks. Following safety evaluation, 34 additional patients were randomized to DCV, ASV, and BMS-791325 150mg BID for 24 (Group 3) or 12 (Group 4) weeks. The primary endpoint was HCVRNA< 25 IU/mL at 12 weeks post-treatment (SVR12). The main outcome of the study for group 2 is shown below in figure 21.
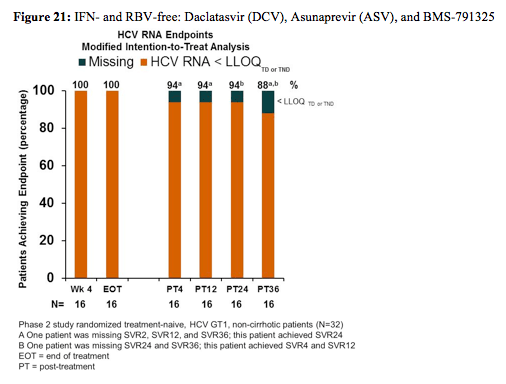
Patients included into this study were mainly GT1a (74%), white race (79%), and IL28B non-CC (70%). Overall, patients achieved SVR4 92% (46/50), SVR12 94% (30/32), and SVR24
94% (15/16). Three failures (Groups 3-4) were observed in patients receiving BMS-791325 150mg (2-viral breakthrough, 1-relapse). No patients discontinued due to adverse events (AEs) related to DCV+ASV+BMS-791325. Most common AEs (≥10% total) were headache, asthenia, and gastrointestinal. Two serious AEs have been reported, both unrelated to DCV+ASV+BMS-791325. No hepatotoxicity or Grade 3/4 elevations of ALT/AST or bilirubin were reported.
In summary the interim results of this ongoing IFN and RBV free phase II trial suggest high SVR rates under treatment of DCV+ASV+BMS-791325 for 12 weeks in HCV GT1 treatment-naïve patients. This study is one of the few which also investigates RBV free regimens which offers hope for patients intolerable to RBV therapy.
EASL: Interim Analysis of an Interferon (IFN)- and Ribavirin (RBV)-Free Regimen of Daclatasvir (DCV), Asunaprevir (ASV), and BMS-791325 in Treatment-Naive, Hepatitis C Virus Genotype 1-Infected Patients - (04/25/13)
EASL: Evaluation of Pharmacokinetic Drug-Drug Interaction (DDI) Between BMS-791325, an NS5B Non-Nucleoside Polymerase Inhibitor, Daclatasvir and Asunaprevir in Triple Combination in HCV Genotype 1-Infected Patients - (04/25/13)
What else was new in the area of HCV?
Obviously, with more and more patients receiving telaprevir or boceprevir based therapy the question remains how can we best treat failures to this first round of DAA based triple therapy. Indeed at EASL a pilot study was presented which looked at the efficacy of combining two new DAAs from different drug classes with a different mode of action and no cross-resistance to HCV protease inhibitors (23). In this study 41 adult HCV GT1-infected patients who had virologic non-response, relapse, or breakthrough during prior treatment with TVR or BOC + pegIFN/RBV were treated with: Daclatasvir (DCV): 60 mg QD NS5A replication complex inhibitor Sofosbuvir (SOF): 400 mg QD NS5B nucleotide inhibitor with or without RBV for 24 weeks of therapy. A total of 41 GT1 non-cirrhotic patients with previous breakthrough (n = 15), relapse (n = 13), or nonresponse (n = 14) to pegIFN/RBV+TVR (n = 33) or BOC (n = 9) (patients who discontinued TVR or BOC due to adverse events were excluded; 1 patient received TVR and BOC) were randomized 1:1 to DCV+SOF with or without RBV for 24 weeks. DCV and SOF were dosed orally at 60mg QD and 400mg QD, respectively. RBV was dosed BID at 1000-1200 mg/d. The primary end point was HCVRNA < 25 IU/mL at 12 weeks post treatment (SVR12). The study design is shown below in figure 22.
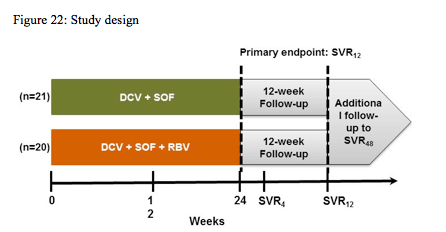
Most patients had HCV GT1a (83%), were IL28B non-CC (98%), and had estimated METAVIR stage ≥F2 (83%). Mean HCVRNA was ≥6 log IU/mL. HCVRNA< 25 IU/mL was achieved in 40/41 patients by week 4 and in all patients by end of treatment. None had breakthrough or relapse and all patients achieved SVR4 (Table). The most common adverse events (>30% total) were fatigue and headache. There were no grade 3-4 hematologic or hepatic laboratory abnormalities. The main findings are shown below in figure 23.
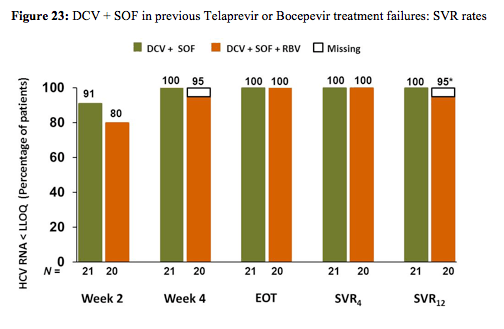
Apparently the combination of DCV + SOF is so potent that it even works without additional ribavirin. These findings are very helpful as they help to envision that there will be potent salvage regimens even in patients failing previous DAA therapy and resistance development.
Another interesting study from the Hannover group investigated the clinical significance of potential DDIs in a real-world cohort of patients receiving PIs for chronic HCV GT 1 infection (24). Between June and November 2011 86 patients with chronic HCV GT 1 who were selected for a PI-based antiviral treatment had a full chart assessment prior to therapy. All drugs were checked for potential DDIs with the approved PIs mainly using different DDI websites and the prescribing information. Risk for DDIs was graded as (1) no significant interaction, (2) potential interaction or (3) should not be coadministered. The 86 patients (mean age 54y) took 74 different comedications. Most common medications were levothyroxine (14%), metformin (12%) and propranolol (12%). Overall, no significant DDIs were expected based on DDI-websites for 65% of the drugs. For 32% DDIs with a PI were considered but were estimated to be tolerable if closely monitored or managed by dose modifications at some point. Only 3% of the drugs were strictly not recommended for
coadministration. In conclusion many patients with chronic HCV GT1 infection were
affected by potential DDIs if treated with a PI. Although number of contraindicated comediactions was low careful current drug intake history appears well advised.
EASL: Sustained Virologic Response With Daclatasvir Plus Sofosbuvir ± Ribavirin (RBV) in Chronic HCV Genotype (GT) 1-Infected Patients Who Previously Failed Telaprevir (TVR) or Boceprevir (BOC) - (04/27/13)
Summary
· Among US-Veterans with HCV infection, decompensated liver disease, anemia, cancer, chronic kidney disease and COPD were the strongest predictors of higher risk of mortality, while HCV treatment was associated with over 50% reduction in mortality in this group. These findings once again underline the importance of HCV therapy to significantly lower risk of mortality in HCV infected individuals.
· The safety profile of TVR or BOC with PEG-IFN/RBV in trials evaluating cirrhotic patients is poor and is associated with increased discontinuation rates due to SAEs compared to those reported in phase III trials. In particular patients with platelets below 110.000/μl and lower albumin levels < 35-40 g/l are at high risk of SAEs and should only be treated in very experienced centers. Cure rates are lower in the context of cirrhotic non-responders but are higher than 50% in previous relapsers. These data strongly suggest that triple therapy must be administered cautiously with intensive safety monitoring in patients with liver cirrhosis.
· High SVR rates from the CONCISE study suggest the potential for GT1, IL28B CC patients with rapid virological response to telaprevir plus peginterferon/ribavirin to shorten treatment duration to12 weeks. This study suggests that markers such as IL28b may still play a role in the future as prediction factors which may allow shortening treatment durations of HCV therapy.
· Findings from the OPTIMIZE study underline that the relative efficacy of TVR bid versus q8h is similar regardless of IL28B genotype and baseline fibrosis stage so that all patient type can benefit from this treatment simplification.
· Faldaprevir plus PegIFN/RBV significantly increased SVR12 rates in HCV GT-1 patients in Europe and Japan compared with PegIFN/RBV alone and was well tolerated. In total, 88% of patients treated with FDV were eligible to stop all treatment at Week 24.
· SMV 150 mg QD + PR in HCV genotype 1-infected, treatment-naïve patients leads to significantly increased SVR12 rates compared with PEG-IFN/RBV alone in around 80-81% of patients. Overall, 85-91% qualified for shortened therapy of 24 weeks. Safety and tolerability was comparable to placebo.
· In simeprevir treated patients a transient and mild bilirubin elevations, not accompanied by increases in ALT, AST or alkaline phosphatase, can be observed. The mechanism is attributed to inhibition of OATP1B1/MRP2 transporters.
· In the NEUTRINO phase III trial sofosbuvir + Peg-IFN + RBV for 12 weeks for genotype 1 resulted in an impressive 90% SVR12 rate, thereby being statistically superior to historical control SVR rate of 60%. Genotype 4, 5, and 6 HCV-infected patients had a 97% SVR12 rate demonstrating good activity in other than GT 1 patients. SOF was well tolerated without any additive effects of SOF to the expected safety profile of Peg-IFN + RBV.
· HCV genotype 2 and 3 are very different to treat and most likely need to be studied separately in the future. Overall, GT 3 infections particularly in the presence of cirrhosis seem more challenging to treat.
· In HCV genotype 2 or 3 patients SOF+RBV demonstrated non-inferiority to PEG-IFN+RGV. Overall, GT2 patients responded extremely well with 97% cure rate under the IFN-free strategy. For genotype 3 patients however the low response rate in particular in cirrhosis patients suggests that either more potent treatment strategies will be needed (for example adding a further DAA) or a possibly a longer treatment duration. SOF+RBV for 12 weeks were well tolerated in treatment-naïve cirrhotic and non-cirrhotic GT2/3 HCV-infected patients. Fewer AEs, discontinuations due to AEs, and Grade ¾ laboratory abnormalities were observed during treatment with SOF+RBV as compared to treatment with PEG+RBV.
· In IFN eligible/intolerable or unwilling patients SOF+RBV for 12 weeks achieved a high SVR12 rate without evidence of resistance and was well tolerated in cirrhotic and non-cirrhotic GT2 and non-cirrhotic genotype 3 HCV-infected patients who do not currently have any treatment options. For cirrhotic genotype 3 patients more potent treatments need to be explored.
· In summary, in the FUSION trial in treatment experienced patients retreatment with sofosbuvir + ribavirin led to SVR rates in HCV genotype 2 infected patients following treatment for 12 or 16 weeks. Extending treatment duration to 16 weeks significantly increased SVR rates in genotype 3 HCV-infected patients, particularly those with cirrhosis.
· In the COMMAND study, daclatasvir in combination with PEG-IFN/RBV achieved higher cure rates than PEG-IFN/RBV alone and allowed shortening of treatment in the majority of patients. Nevertheless, treatment response rates in GT 3 patients are again lower as in GT2 particularly in cirrhosis.
· In the ELECTRON study evaluating IFN-free strategies in HCV GT1 patients adding ledipasvir increased efficacy of SOF + RBV for 12 weeks substantially with 100% SVR12 in both treatment-naïve patients and prior null responders indicating that combination of DAAs may be a very successful strategy to improve response rates in more difficult to treat patient populations. No additional safety or tolerability issues were observed under the addition of ledipasvir.
· In the AVIATOR study SVR rates >90% were achieved in naïve and prior null responder patients with a 3-DAA+RBV regimen, across HCV subtype, IL28B genotype, baseline HCV-RNA or severity of fibrosis. The 12-week duration of therapy in treatment-naïve and previous null-responders seemed to be the best duration choice in the studied populations.
· The IFN and RBV free phase II trial evaluating efficacy and safety of DCV+ASV+BMS-791325 for 12 weeks in HCV GT1 treatment-naïve patients reveals SVR rates above 90%. This study is one of the few which so far has investigated a RBV free regimen which offers hope for patients intolerable to RBV therapy.
· In a small pilot trial the combination of daclatasvir + sofosbuvir in patients who previously failed a telaprevir or boceprevir containing DAA based therapy was very high (even without ribavirin) suggesting good salvage regimens in these patients.
References
1. Erqou1 S, Mohanty A, Butt AA. Predictors of mortality among United States. 48th Annual Meeting of the European Association for the Study of the Liver, April 24-28, 2013, Amsterdam, Netherlands; abstract 453
2. Fontaine H, Hezode C, Dorival C, Larrey D, Zoulim F, de Ledinghen V Canva V, Alric L, Bourlie M, Pol S, Poynard T, Riachi1 G, Bernard PH, Raabe JJ, Gournay J, Metivier S, Pawlotsky JM, Samuel D, Barthe Y, Carrat F, Bronowicki JP, ANRS CO20 CUPIC study group. SVR12 rates and safety of triple therapy including Telaprevir or Boceprevir in 221 cirrhotic non-responders treated in the French early access program (ANRS CO20-CUPIC) 48th Annual Meeting of the European Association for the Study of the Liver, April 24-28, 2013, Amsterdam, Netherlands; abstract 60
3. Maasoumy B, Port K, Markova AA, Calle Serrano B, Sollik L, Kirschner J, Mix C, Manns MP, Wedemeyer H, Cornberg M. Triple therapy for hepatitis C virus (HCV) infection in patients with compensated liver cirrhosis: lessons learned from the first real world experience. 48th Annual Meeting of the European Association for the Study of the Liver, April 24-28, 2013, Amsterdam, Netherlands; abstract 857
4. Petersen J, Matschenz K, Olah K, Unger S, Lorenzen T, Kuhlendahl F, Plettenberg A, Stoehr A, Wolski H, Voelker K, Czaja-Harder C, Buggisch P. Real world experience with triple therapy for HCV GT1 patients in difficult to treat patients:severe adverse events and high rates of virological breakthrough. 48th Annual Meeting of the European Association for the Study of the Liver, April 24-28, 2013, Amsterdam, Netherlands; abstract 453
5. Rutter K, Ferlitsch A, Maieron A, Stättermayer AF, Graziadei I, Dulic M, Gschwantler M, Stauber R, Hofer H, Peck-Radosavljevic M, Vogel W, Trauner M, Ferenci P. Ssfeta of triple therapy with telaprevir or boceprevir in hepatitis C patients with advanced liver disease - predictive factors for sepsis. 48th Annual Meeting of the European Association for the Study of the Liver, April 24-28, 2013, Amsterdam, Netherlands; abstract 65
6. Fried MW, Reddy KR, Di Bisceglie AM, Jensen DM, Jacobson IM,. Sulkowski MS, Terrault NA, Afdhal NH, Gordon SC, Pockros PJ, Kwo PY, Everson GT, Sherman KE, Muir AJ, Pearlman BL, Stewart TG, Vainorius M, Peter JA, Nelson DR, for HCV-TARGET. HCV-TARGET: A longitudinal, observational study of North American patients with chronic hepatitis C (HCV) treated with boceprevir or telaprevir. 48th Annual Meeting of the European Association for the Study of the Liver, April 24-28, 2013, Amsterdam, Netherlands; abstract 818
7. Nelson DR, Poordad F, Feld JJ, Fried MW, Jacobson IM, Pockros PJ, Sulkowski MS, Zeuzem S, Bengtsson L, George S, Friedman MI, on behalf of the CONCISE Study Team. High SVR rates (SVR4) for 12-week total telaprevir combination therapy in IL28B CC treatment-naives and prior relapsers with G1 chronic hepatitis C: Concise interim analyses. 48th Annual Meeting of the European Association for the Study of the Liver, April 24-28, 2013, Amsterdam, Netherlands; abstract 881
8. Buti M, Agarwal K, Horsmans Y, Sievert W, Janczewska E, Zeuzem S, Nyberg L, Brown Jr.RS, Hezode C, Rizzetto M, Parana R, De Meyer S, DeMasi R, Luo D, Witek J. Efficacy of telaprevir dosed twice daily versus every 8 hours by IL28b genotype: Results from the phase III OPTIMIZE study. 48th Annual Meeting of the European Association for the Study of the Liver, April 24-28, 2013, Amsterdam, Netherlands; abstract 798
9. Vierling JM, Zeuzem S, Poordad F, Bronowicki J-P, Manns MP, Bacon BR, Esteban R, Flamm SL, Kwo PJ, Pedicone LD, Deng W, Dutko FJ, DiNubile MJ, Koury KJ, Helmond FA, Wahl J, Bruno S. Safety and efficacy of boceprevir/PEG-Interferon/ribavirin (BOC/P/R) combination therapy for chronic HCV G1 patients with compensated cirrhosis: A meta-analyses of five phase 3 clinical trials. 48th Annual Meeting of the European Association for the Study of the Liver, April 24-28, 2013, Amsterdam, Netherlands; abstract 1430
10. Sclair S, Patel A, Junaidi O, Copado V, Martin P, Rudraraju M, Bhamidimarri KR. Telapprevir or boceprevir therapy in chronic hepatitis C patients with end stage renal disease on hemodialysis is safe, tolerable and feasible. 48th Annual Meeting of the European Association for the Study of the Liver, April 24-28, 2013, Amsterdam, Netherlands; abstract 900
11. Basu PP, Siriki R, Shah NJ, Farhat S, Mittimani K, Atluri S, Rahaman M, Brown Jr. RS. Telaprevir with adjusted dose of ribavirin in naïve CHC-G1: Efficacy and treatment in CHC in hemodialysis population. TARGET C (RCT) 48th Annual Meeting of the European Association for the Study of the Liver, April 24-28, 2013, Amsterdam, Netherlands; abstract 67
12. Ferenci P, Asselah T, Foster GR, Zeuzem S, Sarrazin C, Moreno C, Ouzan D, Maevskaya M, Calinas F, Morano LE, Crespo J, Dufour JF, Bourliere M, Agarwal K, Forton D, Schuchmann M, Zehnter E, Nishiguchi S, Omata M, Stern JO, Datzenko Y, Scherer J, Quinson AM. Falaprevir plus pegylated interferon alfa2a and ribavirin in chronic HCV genotype-1 treatment-naïve patients: Final results from STARTVERSO1, a randomized, placebo-controlled phase III trial. 48th Annual Meeting of the European Association for the Study of the Liver, April 24-28, 2013, Amsterdam, Netherlands; abstract 1416
13. Jacobson I, Dore GJ, Foster GR, Fried MW, Radu M, Rafalskiy VV, Moroz L, Craxì A, Peeters M, Lenz O, Ouwerkerk-Mahadevan S, Kalmeijer R, Beumont-Mauviel M. Simeprevir (TMC435) with peginterferon/ribavirin for chronic HCV genotype-1 infection n treatment-naïve patients: results from QUEST-1, a phase III trial. 48th Annual Meeting of the European Association for the Study of the Liver, April 24-28, 2013, Amsterdam, Netherlands; abstract 1425
14. Manns M, Marcellin P, Poordad FP, Stanislau Affonso de Araujo E, Buti M, Horsmans Y, Janczewska E, Villamil F, Peeters M, Lenz O, Ouwerkerk-Mahadevan S, Kalmeijer R, Beumont-Mauviel M. Simeprevir (TMC435) with peginterferon/ribavirin for chronic HCV genotype-1 infection n treatment-naïve patients: results from QUEST-2, a phase III trial. 48th Annual Meeting of the European Association for the Study of the Liver, April 24-28, 2013, Amsterdam, Netherlands; abstract 1413
15. Lawitz E, Wyles D, Davis M, Rodriguez-Torres M, Reddy KR, Kowdley KV, Svarovskaia E, Jiang D, McNally J, Brainard DM, Symonds WT, McHutchison JG, Nyberg L, Younossi Z. Sofosbuvir + peginterferon + ribavirin for 12 weeks achieves 90% SVR12 in genotype 1, 4, 5, or 6 HCV infected patients: The NEUTRINO study. 48th Annual Meeting of the European Association for the Study of the Liver, April 24-28, 2013, Amsterdam, Netherlands; abstract 1411
16. Gane E, Lawitz E, Rodriguez-Torres M, Gordon S, Dvory-Sobol H, Arterburn S, McNally J, Brainard DM, Symonds WT, McHutchison JG, Sheikh A, Mangia A. Phase 3 randomized controlled trial of all-oral treatment with sofosbuvir + ribavirin for 12 weeks compared to 24 weeks of PEG + ribavirin in treatment naïve GT2/3 HCV-infected patients (FISSION). 48th Annual Meeting of the European Association for the Study of the Liver, April 24-28, 2013, Amsterdam, Netherlands; abstract 5
17. Jacobson I, Yoshida EM, Sulkowski M, Nelson DR, Svarovskaia E, An D, McNally J, Brainard DM, Symonds WT, McHutchison JG, Pianko S, Kowdley KV. Treatment with sofosbuvir + ribavirin for 12 weeks achieves SVR12 of 78% in GT2/3 interferon-ineligible, -intolerant, or unwilling patients: results of the phase 3 POSITRON trial. 48th Annual Meeting of the European Association for the Study of the Liver, April 24-28, 2013, Amsterdam, Netherlands; abstract 61
18. Nelson DR, Feld J, Kowdley KV, Al-Assi MT, Lin M, Mo H, McNally J, Brainard DM, Symonds WT, McHutchison JG, Patel K, Gordon SC. All oral therapy with sofosbuvir - ribavirin for 12 or 16 weeks in treatment experienced GT2/3 HCV-infected patients: results of the phase 3 trial. 48th Annual Meeting of the European Association for the Study of the Liver, April 24-28, 2013, Amsterdam, Netherlands; abstract 6
19. Dore GJ, Lawitz E,Hezode C,Shafran S, Ramji A, Tatum H, Taliani G, Tran A, Brunetto M, Zaltron S, Strasser S, Weis N, Ghesquiere W, Lee S, Larrey D, Pol S, Harley H, George J, Fung S, de L´edinghen V, Hagens P, Cohen D, Cooney E, Noviello S, Hughes S. Daclatasvir combined with peginterferon alfa-2a and ribavirin for 12 or 16 weeks in patients with HCV genotype 2 or 3 infection: COMMAND GT2/3 study. 48th Annual Meeting of the European Association for the Study of the Liver, April 24-28, 2013, Amsterdam, Netherlands; abstract 1418
20. Gane ED, Stedman CA, Hyland RH, Pang PS, Ding X, Symonds WT, McHutchison JG. All-oral sofosbuvir-based 12-week regimes for the treatment of chronic HCV infection: the ELECTRON study. 48th Annual Meeting of the European Association for the Study of the Liver, April 24-28, 2013, Amsterdam, Netherlands; abstract 14
21. Kowdley KV, Lawitz E, Poordad F, Cohen DE, Nelson D, Zeuzem S, Everson GT, Kwo P, Foster GR, Sulkowski M, Xie W, Larsen L, Khatri A, Podsadecki T, Bernstein B. Safety and efficacy of interferon-free regimens of ABT-450/r, ABT-267, ABT-333 +/- ribavirin in patients with chronic HCV GT1 infection: results from the AVIATOR study. 48th Annual Meeting of the European Association for the Study of the Liver, April 24-28, 2013, Amsterdam, Netherlands; abstract 3
22. Everson GT, Sims KD, Rodriguez-Torres M, Hezode C, Lawitz E, Bourliere M, Loustaud-Ratti V, Rustgi V, Schwartz H, Tatum H, Marcellin P, Pol S, Thuluvath PJ, Eley T, Wang X, Huang SP, McPhee F, Wind-Rotolo M, Chung E, Pasquinelli C, Grasela DM, Gardiner DF. Interim analysis of an interferon (IFN)- and ribavirin (RBV)-free regimen of daclatasvir (DCV), asunaprevir (ASV), and BMS-791325 in treatment-naive, hepatitis C virus genotype 1-infected patients. 48th Annual Meeting of the European Association for the Study of the Liver, April 24-28, 2013, Amsterdam, Netherlands; abstract 1423
23. Sulkowski MS, Gardiner DF, Rodriguez-Torres M, Reddy KR, Hassanein T, Jacobson I, Lawitz E, Lok AS, Hinestrosa F, Thuluvath PJ, Schwartz H, Nelson DR, Everson GT, Eley T, Wind-Rotolo M, Huang SP, Gao M, McPhee F, Hernandez D, Sherman D, Hindes R, Symonds W, Pasquinelli C, Grasela DM, AI444040 Study Group. Sustained virological response with daclatasvir plus sofosbuvir ± ribavirin (RBV) in chronic HCV genotype (GT) 1-infected patients who previously failed telaprevir (TVR) or boceprevir (BOC). 48th Annual Meeting of the European Association for the Study of the Liver, April 24-28, 2013, Amsterdam, Netherlands; abstract 1417
24. Maasoumy B, Port K, Markova AA, Calle Serrano B, Sollik L, Manns MP, Cornberg M, Wedemeyer H. Clinical significance of drug-drug-interactions in the era of direct acting antiviral agents against hepatitis C - A real-world experience. 48th Annual Meeting of the European Association for the Study of the Liver, April 24-28, 2013, Amsterdam, Netherlands; abstract 856
|
| |
|
|
|
|
|