| |
Efficacy and safety of simeprevir with PegIFN/ribavirin in
naïve or experienced patients infected with chronic HCV genotype 4
|
| |
| |
Download the PDF here
Download the PDF here
Journal of Hepatology May 2015
Christophe Moreno1, Christophe Hezode2, Patrick Marcellin3, Stefan Bourgeois4, Sven Francque5, Didier Samuel6, Fabien Zoulim7, Jean-Didier Grange8, Umesh Shukla9, Oliver Lenz10, Sivi Ouwerkerk-Mahadevan11, Bart Fevery10, Monika Peeters10, Maria Beumont10, Wolfgang Jessner10
1CUB Hopital Erasme, Universite Libre de Bruxelles, Brussels, Belgium; 2Hopital Henri Mondor, AP-HP, Universite Paris-Est, INSERM U955, Creteil, France; 3Hopital Beaujon, Clichy, France; 4ZNA Campus Stuivenberg, Antwerp, Belgium; 5UZ Antwerpen, Antwerp, Belgium;
6Hopital Paul-Brousse, Centre Hepatobiliaire, Universite Paris Sud, Villejuif, France; 7Hopital de la Croix Rousse, Hepatology Department,
Hospices Civils de Lyon and INSERM U1052, Lyon, France; 8Hopital Tenon, AP-HP, Universite Pierre et Marie Curie, Paris, France;
9Janssen Research & Development, Titusville, NJ, USA; 10Janssen Infectious Diseases BVBA, Beerse, Belgium;
11Janssen Research & Development, Beerse, Belgium
Background & Aims
Simeprevir (SMV) is a once-daily (QD), oral hepatitis C virus (HCV) NS3/4A protease inhibitor approved for treatment of genotype (GT) 1 and GT4 infection. This Phase III, open-label, single-arm study (RESTORE; NCT01567735) evaluated efficacy/safety of SMV with peginterferon-α-2a/ribavirin (PR) in patients with chronic HCV GT4 infection.
Methods
107 patients were included. Treatment-naïve (n = 35) and prior relapse patients (n = 22) received SMV 150 mg QD + PR (12 weeks), followed by PR alone (12 or 36 weeks, response-guided [HCV RNA <25 IU/ml detectable/undetectable at week 4 and <25 IU/ml undetectable at week 12]). Prior non-responders (partial, n = 10; null, n = 40) received SMV/PR (12 weeks), followed by PR for 36 weeks. The primary endpoint was sustained virologic response 12 weeks after end of treatment (SVR12).
Results
Median age: 49.0 years; 28.0% Black/African; 7.5% IL28B CC; 28.8% METAVIR F4. Overall, 65.4% (70/107) of patients achieved SVR12 (82.9% [29/35] treatment-naïve; 86.4% [19/22] prior relapsers; 60.0% [6/10] prior partial responders; 40.0% [16/40] prior null responders). In treatment-naïve and prior relapser patients fulfilling response-guided criteria for 24 weeks of treatment (88.6% [31/35] and 90.9% [20/22]), SVR12 rates were high: 93.5% [29/31] and 95.0% [19/20], respectively. Overall on-treatment failure and relapse rates were 23.4% (25/107) and 14.6% (12/82), respectively. Adverse events (AEs) were mainly grade 1/2; serious AEs were infrequent (4.7%) and considered unrelated to SMV.
Conclusions
Efficacy and safety of SMV 150 mg QD for 12 weeks with PR in treatment-naïve or -experienced patients with chronic HCV GT4 infection were in line with previous reports for HCV GT1 infection.
Introduction
Hepatitis C virus (HCV) genotype (GT) 4 infection is common in the Middle East and sub-Saharan Africa, and accounts for ~90% of HCV infections in Egypt [1]. Its prevalence has increased in several European countries [[2], [3]], which is considered to be largely a consequence of immigration and intravenous drug use [[4], [5]].
Triple therapy with a direct-acting antiviral agent (DAA) in combination with peginterferon plus ribavirin (PR) is the current standard-of-care for chronic HCV GT4 infection. In the USA, sofosbuvir plus PR is the recommended regimen, with simeprevir (SMV) plus PR listed as an alternative option, while in Europe both of these regimens are recommended for GT4 infection [[6], [7]]. SMV is a once-daily (QD), oral HCV NS3/4A protease inhibitor, approved in the European Union (EU), United States and other countries for the treatment of HCV GT1 infection. In the EU, SMV is also approved for the treatment of HCV GT4 infection. In addition to triple therapy, SMV is approved as part of IFN-free combinations.
In vitro, SMV is fully active against HCV GT4, displaying activity similar to that observed against HCV GT1 [8]. The multi-genotypic activity of SMV was confirmed in vivo in a Phase IIa study in treatment-naïve, non-GT1-infected patients in which SMV monotherapy (200 mg QD for 1 week) demonstrated antiviral activity in all HCV genotypes except GT3, with the highest activity observed against GT4 and GT6, followed by GT2 and GT5 [9]. In particular, high antiviral activity against GT4 was observed with a consistent and substantial decline in HCV RNA, which was comparable to that previously observed in studies in HCV GT1-infected patients [[10], [11]]. No difference in efficacy was observed between GT4 subtypes 4a, 4c, and 4d, while there was variability in antiviral activity in GT2-infected patients, possibly caused by the different subtypes included [9]. In the Phase III QUEST-1 and QUEST-2 trials in treatment-naïve patients with HCV GT1 infection, significantly improved sustained virologic response (SVR) rates were observed following the addition of SMV to PR (SVR 12 weeks after planned end of treatment [EOT] [SVR12] rates of 80% and 81% [210/264 and 209/257 patients], respectively), allowing a shorter 24-week overall treatment duration in 85% and 91% of patients (224/264 and 235/257 patients), respectively [[12], [13]]. In treatment-experienced patients, SMV 150 mg QD in combination with PR led to overall SVR12 rates of 61-80% in a Phase IIb study that included prior null responders [14]. In the Phase III ATTAIN study, 70% (101/145) of prior partial responders and 44% (102/234) of prior null responders achieved SVR12 [15], while in the Phase III PROMISE study, 79% (206/260) of SMV/PR-treated prior relapsers achieved SVR12, and 93% (241/260) were eligible to complete treatment at week 24 [16].
Here we report the final results of RESTORE, a Phase III, multicentre, open-label, single-arm study exploring the efficacy and safety of SMV in combination with PR in treatment-naïve and -experienced patients with chronic HCV GT4 infection.
Patients and methods
Patients
Treatment-naïve and -experienced adults aged 18-70 years with chronic HCV GT4 infection and plasma HCV RNA >10,000 IU/ml at screening were considered. Treatment-experienced patients included well-characterised patients with prior relapse or a prior partial or null response to PR treatment.
For all patients, a liver biopsy within 3 years prior to screening, unless contraindicated, was required. In patients with a clinical contraindication for biopsy, non-invasive methods were used (for example, FibroScan, Fibrotest, or Magnetic Resonance-Elastography). For patients with bridging fibrosis or cirrhosis (METAVIR score F3 or F4, respectively), an ultrasound performed within 2 months prior to study enrolment without signs of hepatocellular carcinoma was required.
Exclusion criteria included body mass index (BMI) >32 kg/m2, history or evidence of hepatic decompensation, liver disease of non-HCV aetiology, co-infection with non-GT4 HCV, hepatitis B or HIV infection, previous DAA therapy for HCV, a history of malignancy in the 5 years prior to the study, and evidence of severe retinopathy or a clinically relevant ophthalmological disorder. Patients with platelet count <90,000 cells/mm3 (Black/African <75,000 cells/mm3) were also excluded. Further details are provided in the Supplementary Materials. Patients had to provide written informed consent for participation.
Study design and treatment
This was a Phase III, open-label, single-arm study (NCT01567735; RESTORE) conducted in 8 centres in Belgium and France. The study was approved by the institutional review boards of all participating institutions and was performed in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines.
All patients received SMV 150 mg QD with PR for 12 weeks, followed by PR alone for a further 12 or 36 weeks (Fig. 1). Treatment-naïve patients and prior relapsers, regardless of fibrosis stage, were eligible for a shorter 24-week therapy duration if they met response-guided therapy (RGT) criteria: HCV RNA <25 IU/ml detectable or undetectable at week 4 and <25 IU/ml undetectable at week 12. Patients who did not meet these criteria continued PR until week 48, unless a virological stopping rule was met. Prior null and partial response patients received PR for an additional 36 weeks� fixed duration.
SMV was administered orally as a single 150 mg capsule QD. No SMV dose adjustments were permitted. Peginterferon-α-2a (PegIFNα-2a) 180 μg (Pegasys®, Roche, Europe) was administered as a once-weekly subcutaneous injection and ribavirin (RBV) (Copegus®, Roche, Europe) was given orally as 2 or 3 tablets in the morning and 3 tablets in the evening (total daily dose was dependent on body weight: <75 kg: 1000 mg; ≥75 kg: 1200 mg). Further details are provided in the Supplementary Materials.
Study endpoints
The primary endpoint was SVR12 defined as HCV RNA <25 IU/ml undetectable at actual EOT and HCV RNA <25 IU/ml undetectable or detectable 12 weeks after planned EOT. Key secondary endpoints included SVR 24 weeks after planned EOT (SVR24), undetectable HCV RNA (<25 IU/ml undetectable) and/or HCV RNA levels <25 IU/ml (detectable/undetectable at weeks 4, 12, 24, 36, 48 and follow-up), on-treatment failure (confirmed HCV RNA <25 IU/ml detectable or ≥25 IU/ml at EOT), viral breakthrough (confirmed HCV RNA increase >1 log10 IU/ml after lowest level reached, or confirmed HCV RNA >100 IU/ml in patients whose HCV RNA levels were previously below the lower limit of quantification [LLOQ <25 IU/ml detectable/undetectable] while on study treatment), and viral relapse (HCV RNA <25 IU/ml undetectable at EOT and confirmed detectable HCV RNA levels during follow-up). Primary and secondary objectives were evaluated in patient sub-populations defined by their previous response to PR treatment.
Study assessments
Samples for plasma HCV RNA determination were collected at screening, baseline, on days 3 and 7, at weeks 2, 3, 4, 8, 12, 16, 20, and 24 (all patients) and at weeks 28, 36, 42, and 48 (patients who continued PR until week 48). HCV RNA samples were also collected during post-therapy follow-up (weeks 28, 36, and 48 for patients receiving PR until week 24; weeks 52, 60, and 72 for those receiving PR until week 48). Plasma HCV RNA was determined at a central laboratory using the Roche COBAS® TaqMan® v2.0 HCV assay system (LLOQ: 25 IU/ml; limit of detection: 12 IU/ml for GT4). Sequencing of the HCV NS3/4A protease domain was performed on baseline samples and post-baseline in patients not achieving SVR at selected time points based on HCV RNA changes using standard population-based sequencing.
Adverse events (AEs) were monitored throughout the study and during follow-up. AEs were graded by investigators according to the World Health Organization (WHO) grading scale. Laboratory abnormalities were graded according to WHO criteria. Further details are provided in the Supplementary Materials.
Statistical analysis
Analyses were performed on the intent-to-treat (ITT) population (i.e., all patients who took at least one dose of study medication). No formal power calculation was performed as no formal hypothesis was defined. Assuming 100 patients with chronic HCV GT4 infection were enrolled, the one-sided width of the 95% two-sided confidence interval (CI) around the SVR rate was at the maximum 10%.
95% CIs were constructed around the observed response rates both in the overall population and in the four sub-populations based on response to prior treatment. Subgroup analyses were performed based on previous treatment response and GT4 subtype, METAVIR fibrosis score and IL28B genotype. Time to achieve undetectable HCV RNA was estimated using Kaplan-Meier plots. Change in log10 HCV RNA from baseline at all time points, demographic and baseline characteristics and the incidence of AEs/laboratory abnormalities were analysed descriptively.
Results
Patient disposition
The study was conducted between 16 February 2012 and 20 March 2014. A total of 136 patients from 8 centres in France and Belgium were screened. Of these, 107 received treatment and were included in the ITT population (35 treatment-naïve, 22 prior relapsers, 10 prior partial responders and 40 prior null responders) (Fig. 2). At the time of the final analysis, 103 patients (96.3%) had completed the study and 4 (3.7%) had discontinued early (3 lost to follow-up and 1 withdrew consent).
Most patients completed SMV treatment (98/107 [91.6%]). Nine patients (8.4%) discontinued SMV treatment; 8 (7.5%) due to meeting a virological stopping rule and 1 (0.9%) due to an AE (see Safety section).
Patient demographic and baseline characteristics are shown in Table 1. No patients with cirrhosis (METAVIR score F4) had significant thrombocytopenia at baseline.
Efficacy
Virological response
The overall rate of SVR12 was 65.4% (70/107; 95% CI: 56.4-74.4). The SVR12 rate was 82.9% (29/35) in treatment-naïve patients, 86.4% (19/22) in prior relapsers, 60.0% (6/10) in prior partial responders and 40.0% (16/40) in prior null responders (Table 2). All patients who achieved SVR12 also achieved SVR24 (70/70 [100%]).
Table 2Primary and key secondary efficacy endpoints for all patients and by prior treatment response.
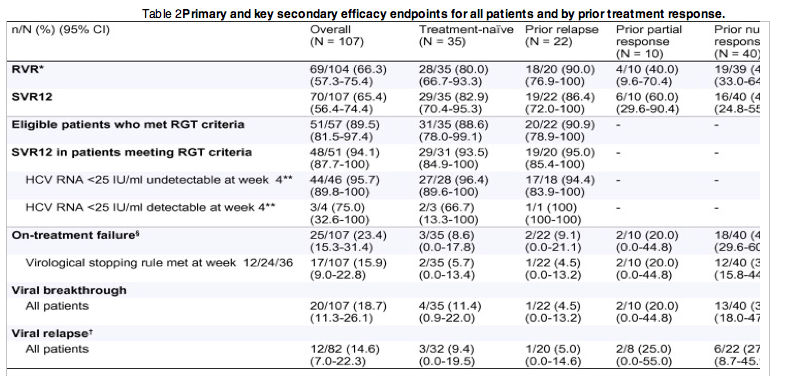
3 patients did not have a week 4 HCV RNA assessment available.
Patients with a week 4 HCV RNA assessment available (1 patient did not have a week 4 HCV RNA assessment available; the patient achieved SVR12).
HCV RNA confirmed detectable at EOT.
Among patients with undetectable HCV RNA at EOT and at least one follow-up HCV RNA measurement.
CI, confidence interval; EOT, end of treatment; RGT, response-guided therapy (criteria: HCV RNA week 4 <25 IU/ml detectable or undetectable at week 4 and <25 IU/ml undetectable at week 12); RVR, rapid virologic response (HCV RNA week 4 undetectable); SVR12, sustained virologic response at 12 weeks (HCV RNA <25 IU/ml undetectable at the EOT and <25 IU/ml detectable or undetectable 12 weeks after the planned EOT).
Overall, 69 patients (66.3%; 95% CI: 57.3-75.4) achieved rapid virologic response (RVR; defined as HCV RNA <25 IU/ml undetectable at week 4). More specifically, 80.0% (28/35) of treatment-naïve, 90.0% (18/20) of prior relapsers, 40.0% (4/10) of prior partial responders and 48.7% (19/39) of prior relapsers had RVR (Table 2). All 4 prior partial responders with RVR achieved SVR12, while 13/19 null responders with RVR achieved SVR12. Among the 6 null responders with RVR who did not achieve SVR12, 5 had METAVIR score F4 (F3, n = 1) and all 6 had IL28B non-CC genotype. At week 4, the majority of patients (85.6% [89/104]) had HCV RNA <25 IU/ml undetectable/detectable (88.6% [31/35] treatment-naïve; 95.0% [19/20] prior relapser; 90.0% [9/10] prior partial responder; 76.9% [30/39] prior null responder). 14.4% (15/104) of patients had HCV RNA ≥25 IU/ml at week 4 (11.4% [4/35] treatment-naïve; 5.0% [1/20] prior relapser; 10.0% [1/10] prior partial responder; 23.1% [9/39] prior null responder); none of these achieved SVR12.
Overall, 31/35 (88.6%; 95% CI: 78.0-99.1) treatment-naïve patients and 20/22 (90.9%; 95% CI: 78.9-100) prior relapsers met RGT criteria and were eligible to complete PR therapy at week 24; of these, 29 (93.5%) and 19 (95.0%), respectively, achieved SVR12 (Table 2). Among patients who met RGT criteria, 95.7% (44/46) of those with HCV RNA <25 IU/ml undetectable at week 4 achieved SVR12 compared with 75.0% (3/4) of those with HCV RNA <25 IU/ml detectable at week 4. Of the 11 patients with METAVIR score F4 who were eligible for RGT, 9 (81.8%) met the criteria; of those, 8 (88.9%) achieved SVR12.
Race did not have an effect on SVR, with a total of 76.7% (23/30) of Black/African patients achieving SVR12 compared with 61.0% (47/77) of White patients (Table 3). Similarly, age did not have an effect on SVR12 rates, whereas a BMI ≥30 kg/m2 was associated with a lower response. Overall, SVR12 rates were similar across the different HCV GT4 subtypes, with slightly lower rates observed among patients with GT4d (Table 3). SVR12 was achieved by 8/8 (100%) patients with IL28B CC genotype compared to 40/61 (65.6%) and 22/37 (59.5%) of patients with CT and TT genotypes, respectively. The SVR12 rate in patients with METAVIR score F4 was 46.7% (14/30); of note, the majority of these patients [63%; 19/30] were prior partial and null responders). The SVR12 rate in patients with METAVIR score F0-F2 was 76.3% (45/59) compared with 66.7% (10/15) in patients with F3; 37% (22/59) of patients with METAVIR F0-F2 and 13% (2/15) of those with F3 were prior partial or null responders (Table 3).
Treatment failure
On-treatment virological failure (i.e. confirmed detectable HCV RNA at EOT) occurred in 23.4% (25/107; 95% CI: 15.3-31.4) of patients (Table 2). Viral relapse was observed in 12/82 (14.6%; 95% CI: 7.0-22.3) patients. Highest relapse rates were observed in prior null responders (27.3% [6/22] vs. 9.4% [3/32], 5.0% [1/20] and 25.0% [2/8] in treatment-naïve, prior relapsers and prior partial responders, respectively). No patient who met RGT criteria experienced on-treatment failure, while 3 (2 treatment-naïve patients and 1 prior relapser) had viral relapse.
NS3 sequence information was available for 32/37 SMV-treated patients with treatment failure, of whom 87.5% (28/32) had emerging NS3 mutations at the time of failure at NS3 amino acid positions 80, 122, 155, 156 and/or 168. Mutations at position 168 were the most frequently observed (75.0% [24/32]) at the time of failure: mainly D168V alone and D168E alone or combined with mutations at position 80.
Safety
Adverse events
The majority of AEs during the SMV/PR treatment phase, summarised in Table 4, were either worst grade 1 or 2 (91.6% [98/107]). Serious AEs were infrequent (reported in only 5 [4.7%] patients) and all were considered unrelated to SMV (Table 4). There were no fatal AEs and no incidences of hepatic decompensation.
One patient stopped SMV due to a medication error (valium overdose), not related to study treatment; the same patient had previously experienced bradycardia and hypoglycaemia, which led to interruption of SMV. No other AEs led to SMV interruption or discontinuation. The most frequent AEs (occurring in >25% of patients) were influenza-like illness, asthenia and fatigue. Among patients with METAVIR score F4, one grade 3/4 AE (neutropenia) was reported, while 2 patients experienced serious AEs (bradycardia, hypoglycaemia and valium and methadone overdoses, and anaemia, respectively).
Pruritus was reported in 20.6% (22/107) of patients during the SMV/PR phase, while 14.0% (15/107) and 10.3% (11/107) of patients experienced rash (any type) and anaemia, respectively, during the same period. There were no grade 3/4 pruritus or rash AEs reported, while there was a single grade 3 anaemia AE. Photosensitivity was reported in 2 patients; both AEs were grade 1, considered possibly related to SMV, and did not lead to a change in dose of any study drugs. Dyspnoea was reported in 11.2% (12/107) of patients (no grade 3/4 AEs), and neutropenia in 4.7% (5/107) (1 grade 3 AE [0.9%]; 1 grade 4 AE [0.9%]).
Laboratory abnormalities
Mean values for bilirubin, haemoglobin, neutrophils and liver transaminases during the entire treatment phase are shown in Supplementary Fig. 1. No grade 4 hyperbilirubinemia, decreases in haemoglobin or liver transaminase changes were reported. There were 5 (4.7%) cases of grade 4 changes in neutrophils in the SMV/PR phase (see also Supplementary Materials).
Discussion
Limited data are available to guide clinical decision-making in patients with chronic HCV GT4 infection. HCV GT4 is considered difficult to cure compared to GT2 and GT3; however, GT4 has not been extensively studied. Previously, sofosbuvir plus PR led to a cure rate of 96% in patients with GT4 in the NEUTRINO study; however, the patient numbers with GT4 in that study were small (n = 28) and treatment-experienced patients were not included [17]. In this open-label, Phase III study, the overall SVR12 rate observed in patients with HCV GT4 infection was 65% (70/107) and in treatment-naïve and prior relapse patients was 83% (29/35) and 86% (19/22), which is in line with SVR12 rates observed in Phase III studies of SMV in combination with PR in patients with HCV GT1 infection [[12], [13], [16]]. It is also worth noting that the SVR12 rate in prior null responders in this study (40% [16/40]) was consistent with results in HCV GT1-infected null responder patients treated with SMV/PR; in the Phase IIb ASPIRE study, SVR12 rates of 41-59% were observed with SMV/PR in null responders [14], while in the Phase III ATTAIN study, 44% (102/234) of null responders treated with SMV/PR achieved SVR12 [18]. In a study in which HCV GT4-infected patients who had failed previous PR treatment (relapsers, non-responders, treatment failures) were re-treated with PR alone, 28% (19/67) achieved SVR [19]; in RESTORE, 56.9% (41/72) of treatment-experienced patients achieved SVR with SMV/PR. Results from the RESTORE study are clinically highly relevant, as evidenced by the current EASL guidelines, which recommend the use of SMV in combination with PR in GT4 treatment-naïve and -experienced patients [7], as well as the AASLD guidelines which recommend this regimen as an alternative for treatment-naïve patients who are eligible to receive interferon [6]. Simeprevir has also been approved in the EU for the treatment of HCV GT4 infection, both in combination with PR and in interferon-free regimens in combination with sofosbuvir, with or without RBV [20].
Several studies evaluating the efficacy of 48 weeks of PR treatment in patients with HCV GT4 infection have shown SVR rates ranging from 45-68% with PegIFNα-2b [[21], [22]] to 53% with PegIFNα-2a [23], with strong evidence that PR treatment for 48 weeks is more effective than 24 weeks of therapy [[5], [24]]. In the RESTORE study, which to our knowledge is the largest study to date that has evaluated the efficacy of a protease inhibitor-based regimen in combination with PR in an HCV GT4 population, an RGT strategy was employed in treatment-naïve patients and prior relapsers to allow shortening of PR treatment to 24 weeks based on virological response (HCV RNA <25 IU/ml detectable or undetectable at week 4 and <25 IU/ml undetectable at week 12). Most eligible patients (89.5% [51/57]) met RGT criteria; 93.5% (29/31) of treatment-naïve patients and 95.0% (19/20) of prior relapsers who met RGT criteria achieved SVR12. These results support the use of an RGT-based approach to individualise the duration of treatment in HCV GT4-infected patients. Shortening of treatment duration in these patients may be beneficial, as it would reduce overall drug exposure and minimise therapy costs. Previous data in GT4-infected patients treated with PegIFNα-2b and undetectable HCV RNA at weeks 4 and 12 showed that similar outcomes were obtained with shorter (24 or 36 weeks) regimens compared with 48 weeks of PR treatment [25]. In RESTORE, high SVR12 rates (88.9% [8/9]) were also achieved in patients with METAVIR score F4 who met RGT criteria (81.8% [9/11]), indicating that this strategy may be successfully applied to patients with cirrhosis; however, due to the small patient numbers in this group (n = 11), these results should be verified in a larger study.
Consistent with experience in HCV, GT1-infected patients treated with SMV/PR, subgroup analyses in this study revealed little impact of unfavourable baseline characteristics on SVR12 rates; however, it should be noted that the patient numbers included in these analyses were small and may therefore limit the interpretation of these findings. The reason for the slightly lower SVR12 rates among patients with HCV GT4d compared with other GT4 subtypes is uncertain, and this may be a chance finding due to the low number of patients in the GT4d subgroup. Of note, a higher proportion of HCV GT4d-infected patients had cirrhosis compared to GT4a and GT4other patients (36.0% vs. 28.9 and 24.3%, respectively). No difference in in vitro and in vivo susceptibility to SMV between GT4d and other GT4 subtypes has been observed [[9], [26]]. The lower proportion of prior null responders among Black/African patients compared with White patients (23.3% vs. 42.9%, respectively) may explain the slightly higher SVR12 rates observed in Black participants in the study. Although all patients with a prior partial or null response had non-CC IL28B genotypes, this did not greatly affect SVR12 rates, suggesting that SMV may help overcome the association between IL28B genotype and treatment response [27]. GT4-infected prior relapsers with METAVIR F4 score achieved SVR12 at a comparable rate to their GT1-infected counterparts (77.8% [7/9] vs. 74.4% [29/39] in the PROMISE trial) [16]. Similarly, in the present study, cirrhotic null responders with GT4 infection achieved SVR12 rates (35.7% [5/14]) that were comparable to those in GT1-infected patients (25-31% [[14], [18]]). As these subgroup analyses comprised only a small number of patients, the results should be verified in a larger population.
As expected, baseline Q80K polymorphism was not present in GT4 sequences in the LANL HCV sequence database (data not shown). Furthermore, the frequency and type of emerging mutations were consistent with those seen in Phase III studies in SMV-treated patients with HCV GT1 infection [[12], [13], [16]].
The safety and tolerability profile of SMV in HCV GT4-infected patients was in line with that reported in Phase III SMV trials in GT1-infected patients. AEs were mainly Grade 1 or 2, with few serious AEs reported and no deaths. The safety profile was also favourable among patients with cirrhosis. Only a single patient discontinued SMV due to an AE, which was considered unrelated to study treatment (valium overdose). The most common AEs seen during SMV/PR treatment were influenza-like illness, asthenia and fatigue, while no unexpected AEs were reported. Only 2 cases of mild photosensitivity were reported despite no formal recommendation for sun-protective measures. Changes in laboratory parameters were consistent with those observed in the Phase III SMV studies.
In the RESTORE study, the most prevalent HCV GT4 subtypes in Europe and Egypt [1] were adequately represented, with 42.5% and 23.6% of participants having GT4a and GT4d infections, respectively. In addition, a further strength of this study was the inclusion and evaluation of patients according to prior treatment response (although the numbers of prior relapsers and partial responders were limited). Importantly, difficult-to-treat patients with cirrhosis were well represented. In addition, similar to SMV GT1 studies, treatment-naïve and prior relapse patients were eligible for shortened treatment duration.
Study limitations included the lack of a control arm; the study design was guided by the fact that SMV has demonstrated similar activity in GT4-infected patients as in those with GT1 infection [[9], [28]]; therefore, adding SMV to PR was expected to increase SVR rates and shorten therapy duration in this population in line with results of Phase III studies in patients with HCV GT1 infection [[12], [13], [16]]. Another potential limitation is that this study was not conducted in the Middle East, where HCV GT4 infections are associated with a particularly high disease burden [5]. However, there is no obvious virological reason why results from the study cannot be extrapolated to patients from the Middle East. The increasing prevalence of HCV GT4 infections in Europe largely reflects immigration patterns, and HCV GT4 subtype distribution in RESTORE (which covered a broad variety of GT4 subtypes) was representative of HCV GT4 subtype distribution in the Middle East, with HCV GT4a and 4d overall being the most prevalent [[1], [4], [29]].
The effectiveness of protease inhibitor-based regimens in combination with PR may be limited in patients who are non-responders to previous PR therapy, as IFN sensitivity is a key determinant of treatment success [30]. Higher SVR rates are observed in these patients with IFN-free combinations. Based on results in HCV GT1-infected patients, sofosbuvir in combination with daclatasvir is recommended in the current EASL guidelines for HCV GT4-infected treatment-experienced patients [7], and, as previously mentioned, the combination of SMV and sofosbuvir is approved in Europe for GT4-infected patients regardless of prior treatment history [20]. In addition, recent results have shown that the combination of sofosbuvir and ledipasvir for 12 weeks was efficacious and well tolerated in GT4-infected treatment-naïve and -experienced patients [31]. Other IFN-free regimens that have shown significant efficacy in HCV GT1-infected null responders (suggesting that these results could be extrapolated to GT4 patients) include a combination of the HCV protease inhibitor ABT-450, ritonavir, the NS5A inhibitor ombitasvir (ABT-267), the HCV polymerase inhibitor dasabuvir (ABT-333) and RBV [32] and daclatasvir combined with asunaprevir [33].
In conclusion, the RESTORE study supports the efficacy of SMV 150 mg QD in combination with PR in treatment-naïve or -experienced patients with HCV GT4 infection. The majority of treatment-naïve and prior relapse patients, including those with METAVIR score F4, were able to complete treatment at 24 weeks, achieving high SVR12 rates. Patients with prior null response achieved SVR12 rates that were comparable to those reported in GT1 infection. The SMV in combination with PR regimen was safe and well tolerated, further supporting its use in patients with HCV GT4 infection.
|
|
| |
| |
|
|
|